Clear Sky Science · en
Optimizing cross-domain transfer for universal machine learning interatomic potentials
Smarter Simulations for Real Materials
Designing new batteries, catalysts, and electronic materials increasingly depends on computer simulations that track atoms in motion. The most trusted simulations, based on quantum mechanics, are extremely accurate but far too slow for exploring millions of candidate materials. Faster machine‑learning models can mimic quantum calculations, yet they often work only in narrow situations—for example, just crystals or just molecules. This paper proposes a way to build one universal model, called SevenNet‑Omni, that stays accurate across many kinds of materials, from metal surfaces and molecular liquids to porous frameworks, while still running fast enough for large‑scale discovery.
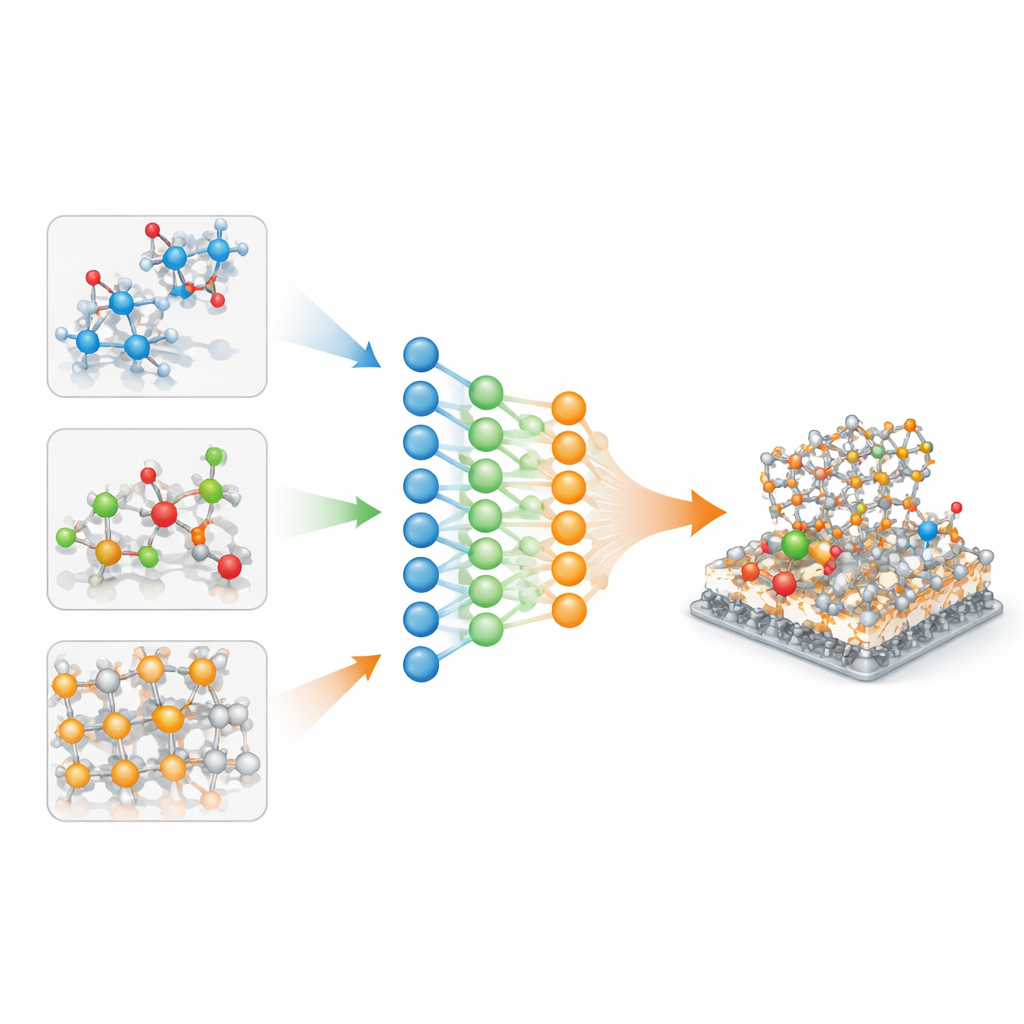
Why Today’s Atomic Models Don’t Travel Well
Current machine‑learning interatomic potentials are usually trained on a single, carefully curated database: one for inorganic crystals, another for drug‑like molecules, yet another for catalytic surfaces. Each database is built with its own quantum‑chemistry settings, so the underlying energy landscapes differ in subtle, non‑linear ways. Simply stitching these data together—perhaps by shifting or scaling energies—adds noise and leads to models that fit their home domain well but fail when asked to describe unfamiliar chemistries or slightly different quantum methods. As materials problems increasingly mix domains, such as molecules reacting on solid surfaces in solution, this lack of transferability has become a serious bottleneck.
A Shared Backbone with Gentle Specialization
The authors address this by treating each database as its own “task” in a single, multi‑task neural network. Inside the model, one set of parameters forms a shared backbone, capturing general rules of atomic bonding, while smaller, task‑specific parameters fine‑tune the behavior for particular datasets. A mathematical analysis shows that, if the task‑specific parts grow too large, the model effectively memorizes each database and forgets how to generalize. To prevent this, the authors apply selective regularization: they directly penalize large task‑specific parameters but leave the shared backbone free to grow as needed. This nudges the network to explain as much as possible through common physics, using only modest corrections for each domain.

Bridging Distant Worlds with a Few Key Examples
Even with regularization, some regions of chemical space appear only in one database, so the shared backbone receives no guidance there. To fix this, the team introduces a “domain‑bridging set.” They carefully select a tiny fraction—about one in a thousand—of configurations from several databases and recompute them using a common quantum‑mechanical setup. These bridging structures act like bilingual phrases in a language textbook: they directly connect how two different quantum methods describe the same atomic scene. When included in training, they strongly tighten the link between tasks, aligning the energy landscapes without the need for brute‑force re‑calculation of everything. Systematic tests show that regularization and the bridging set reinforce each other, improving performance beyond what either can do alone.
Building and Testing a Universal Atomic Engine
Based on these ideas, the authors train SevenNet‑Omni on 15 public datasets comprising about 242 million atomic structures, covering molecules, crystals, catalysts, metal–organic frameworks, and multiple levels of quantum theory. They then benchmark the model in both familiar and challenging situations: crystal stability, grain boundaries in metals, defects in steels, torsion barriers in drug‑like molecules, hybrid organic–inorganic perovskites, adsorption in porous frameworks relevant to carbon capture, and reactions on metal surfaces important for hydrogen and carbon‑dioxide conversion. Across these tests, SevenNet‑Omni often matches or surpasses specialized models trained for a single domain, and it maintains “chemical accuracy” for many reaction and adsorption energies. It also accurately reproduces results from an expensive quantum method (r²SCAN) by learning how that method relates to cheaper, more abundant data.
What This Means for Discovering New Materials
For non‑experts, the key message is that SevenNet‑Omni behaves much like a seasoned scientist who has worked in many subfields. Instead of overfitting to one narrow problem, it learns broad chemical principles and applies them flexibly to new situations, from gas capture in porous solids to reactions on metal electrodes. The paper shows that this is possible by carefully sharing information between datasets while lightly constraining their differences, and by adding a small number of carefully chosen “translation” examples between quantum methods. As larger and more diverse databases continue to appear, this training strategy offers a scalable path toward truly universal, trustworthy atomic models that can accelerate discovery across chemistry, physics, and materials science.
Citation: Kim, J., You, J., Park, Y. et al. Optimizing cross-domain transfer for universal machine learning interatomic potentials. Nat Commun 17, 3432 (2026). https://doi.org/10.1038/s41467-026-70195-8
Keywords: machine-learning interatomic potentials, multi-domain materials modeling, transfer learning, universal atomistic potential, materials discovery