Clear Sky Science · sv
Vätskekristallprediktor: en maskininlärningsplattform för klassificering och fasövergångsprognoser
Varför det är viktigt att förutsäga mjuk materia
Från telefonskärmar till smarta fönster och mjuka robotar bygger många moderna teknologier på vätskekristaller — material som flyter som vätskor men ändå behåller viss ordning som i fasta ämnen. Att designa nya vätskekristaller är fortfarande till stor del en metod baserad på försök och misstag, eftersom det är svårt att förutsäga vid vilka temperaturer de växlar mellan ordnade och oordnade tillstånd. Denna studie presenterar en öppen plattform baserad på maskininlärning som hjälper forskare att förutspå när en kandidatmolekyl smälter in i en vätskekristallfas och när den förlorar sin ordning helt, vilket gör det lättare att ta fram bättre material för framtida enheter.
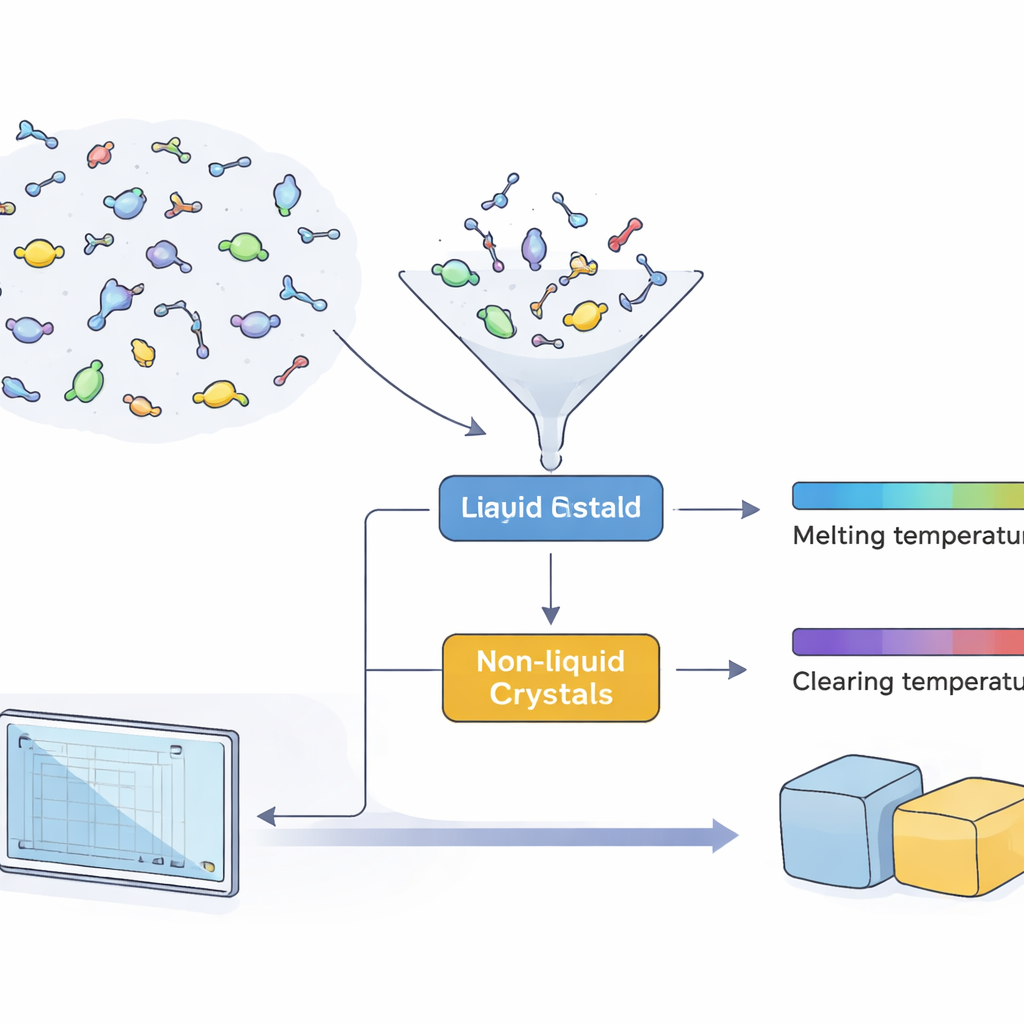
Vad som gör dessa speciella vätskor användbara
Vätskekristaller intar en märklig mellanposition mellan fast och flytande. Deras molekyler — ofta stavformade, skivformade eller böjda — tenderar att peka i liknande riktningar och skapar faser som är ordnade men flytande. För praktisk användning måste denna ordning vara stabil inom ett visst temperaturområde, vanligtvis definierat av två viktiga punkter: smälttemperaturen, där en fast kristall först blir en vätskekristallfas, och klarningstemperaturen, där den fasen slutligen övergår till en vanlig vätska. Att känna till dessa två temperaturer i förväg låter ingenjörer avgöra om ett material fungerar i till exempel en sensorsom arbetar i rumstemperatur, en varm bildskärm i en bil eller en medicinsk apparat.
Att lära datorer känna igen lovande molekyler
Forskarna sammanställde och rensade först en stor, offentlig datamängd med 11 335 organiska molekyler. Bland dessa är 1 256 kända vätskekristaller som spänner över de tre huvudfamiljerna — stavlika, diskotiska och bent‑core — medan resten är icke‑vätskekristallföreningar hämtade från många delar av kemin. Med detta material tränade och jämförde de flera maskininlärningsmodeller för att skilja vätskekristallmolekyler från allt annat. Genom att kombinera tre kompletterande modeller i ett "majoritetsröstnings"‑schema som favoriserar att kalla en kandidat för vätskekristall snarare än att missa den, återfick deras klassificerare nästan alla kända vätskekristaller i ett oberoende test, med särskilt stark prestation för de mer komplexa diskotiska och bent‑core‑typerna.
Att förutse nyckeltemperaturer utifrån molekylform
När en molekyl väl taggats som en sannolik vätskekristall är nästa utmaning att förutsäga dess smält‑ och klarningstemperaturer. För att tackla detta jämförde teamet traditionella algoritmer som bygger på förberäknade fingeravtryck av molekylstrukturen med en nyare metod som behandlar varje molekyl som en graf av atomer och bindningar. För smälttemperaturer gav en hybridmodell som blandar en random‑forest‑regressor med ett grafneuralt nätverk bäst total noggrannhet och hanterade framgångsrikt de olika beteendena hos stavlika, diskotiska och bent‑core‑material. För klarningstemperaturer generaliserade den grafbaserade modellen bäst på egen hand, troligen eftersom denna varmare övergång beror mer på molekylens globala form och kopplingar än på lokala detaljer.
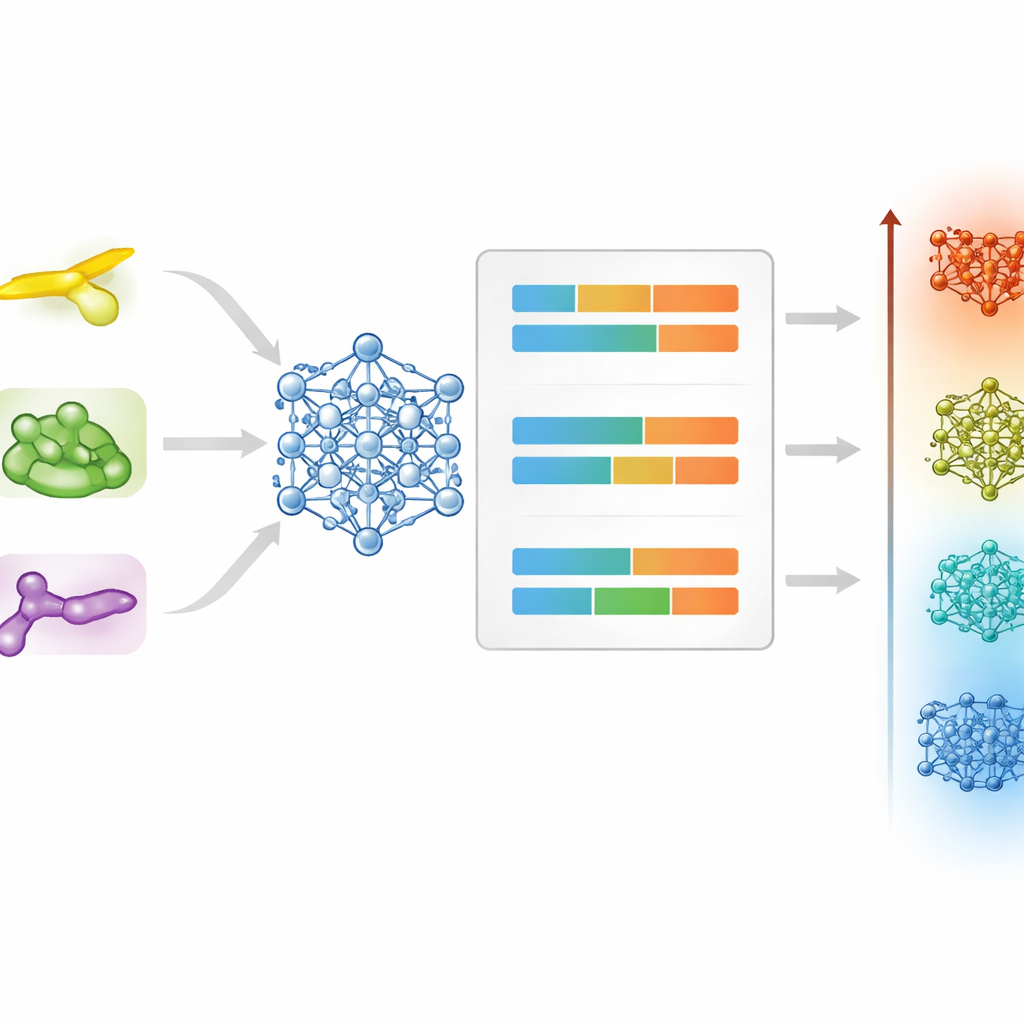
Att se subtila mönster och förstå misslyckanden
Ett strängt test av vilket prediktivt verktyg som helst är om det kan fånga ömtåliga mönster som kemister känner igen från erfarenhet. Här reproducerade modellerna inte bara typiska temperaturintervall utan speglade också så kallade udda–jämn‑effekter, där det att lägga till eller ta bort en enda kol i en sidokedja får övergångstemperaturerna att oscillerande ändras. Författarna granskade också fall där förutsägelserna var fel med mer än cirka 30 grader. Dessa problematiska molekyler hade ofta mycket böjda ryggrader, skrymmande skivformade kärnor eller ovanliga substituenter som flera fluoratomer — alla faktorer som kan förändra hur molekyler packas tillsammans på sätt som nuvarande data och deskriptorer inte fullt ut fångar. Denna analys pekar på var ytterligare experiment och förfinade egenskaper kan förbättra verktyget ytterligare.
En ny genväg för att designa framtida material
All data, modeller och ett användarvänligt webbgränssnitt görs öppet tillgängliga som Liquid Crystal Predictor. En forskare kan nu skissa eller specificera en molekyl, låta plattformen avgöra om den sannolikt bildar en vätskekristallfas och uppskatta de temperaturer vid vilka den kommer att framträda och försvinna — utan att behöva djup expertis i maskininlärning. Även om noggrannheten fortfarande är lägre för vissa underrepresenterade molekylfamiljer erbjuder systemet redan en kraftfull vägledning för att sålla kandidater före syntes och testning. Med tiden, när mer exotiska strukturer och bättre strukturella deskriptorer läggs till, skulle verktyg som detta kunna omvandla sökandet efter avancerade vätskekristallmaterial från en konst till en datadriven, kollaborativ vetenskap.
Citering: Wu, H., Patel, H., Xiang, Y. et al. Liquid crystal predictor: a machine learning platform for classification and phase transition forecast. npj Soft Matter 2, 11 (2026). https://doi.org/10.1038/s44431-026-00020-1
Nyckelord: vätskekristaller, maskininlärning, fasövergångar, materialdesign, mjuk materia