Clear Sky Science · it
Predittore di cristalli liquidi: una piattaforma di machine learning per la classificazione e la previsione delle transizioni di fase
Perché prevedere la materia soffice è importante
Dai display dei telefoni alle finestre intelligenti e ai robot morbidi, molte tecnologie moderne si basano sui cristalli liquidi—materiali che scorrono come liquidi ma conservano parte dell’ordine tipico dei solidi. Progettare nuovi cristalli liquidi è ancora in larga parte un processo di tentativi ed errori, perché è difficile prevedere le temperature a cui passano tra stati ordinati e disordinati. Questo studio presenta una piattaforma aperta basata su machine learning che aiuta gli scienziati a prevedere quando una molecola candidata si trasformerà in un cristallo liquido e quando perderà completamente il suo ordine, facilitando l’invenzione di materiali migliori per i dispositivi del futuro.
Perché questi liquidi speciali sono utili
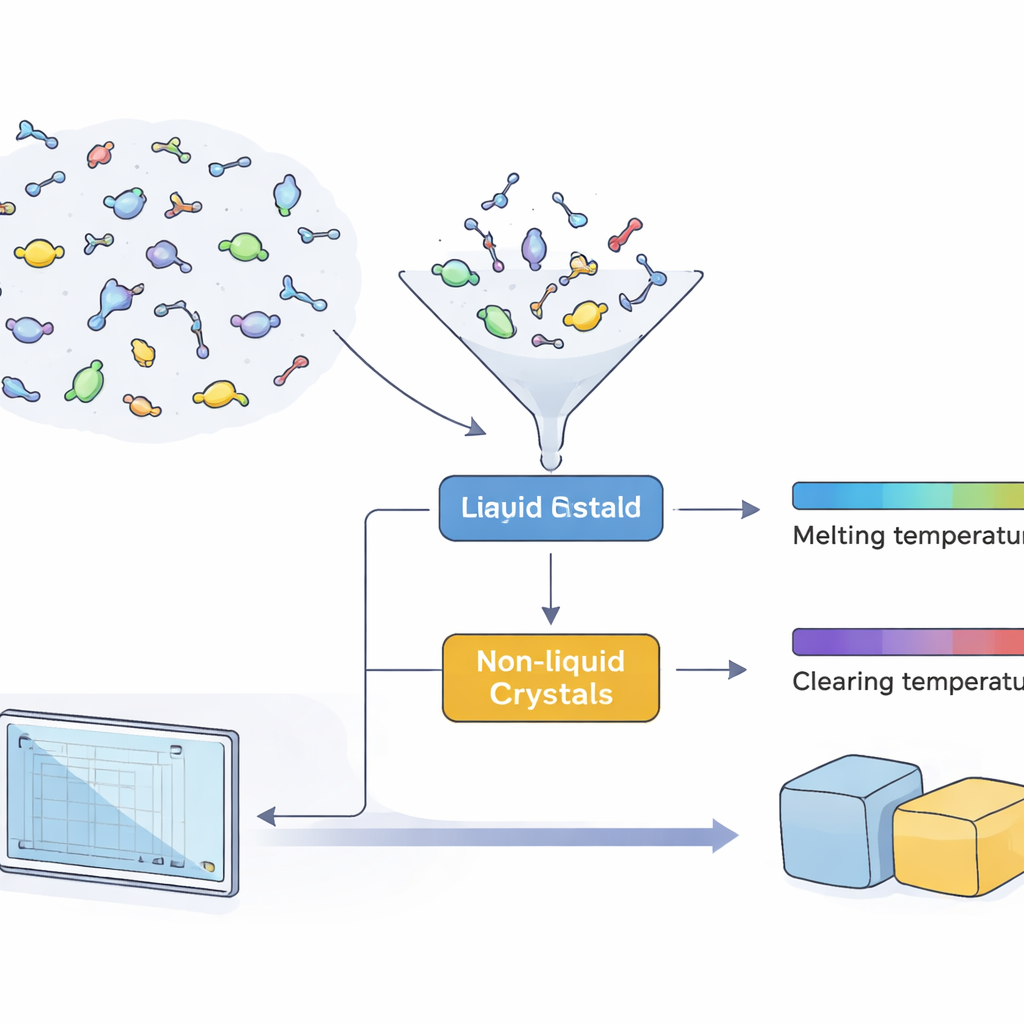
I cristalli liquidi occupano un curioso spazio intermedio tra solido e liquido. Le loro molecole—spesso a forma di bastoncino, disco o ad angolo—tendono ad allinearsi in direzioni simili, creando fasi che sono ordinate ma fluide. Per qualsiasi applicazione pratica, questo ordine deve essere stabile in una specifica finestra di temperatura, solitamente definita da due punti chiave: la temperatura di fusione, in cui un cristallo solido diventa per la prima volta una fase a cristallo liquido, e la temperatura di chiarimento, dove quella fase si trasforma finalmente in un liquido ordinario. Conoscere in anticipo queste due temperature permette agli ingegneri di stabilire se un materiale funzionerà, per esempio, in un sensore a temperatura ambiente, in un display di un’auto esposto a temperature elevate o in un dispositivo medico.
Insegnare ai computer a riconoscere molecole promettenti
I ricercatori hanno innanzitutto raccolto e pulito un ampio set di dati pubblico di 11.335 molecole organiche. Di queste, 1.256 sono cristalli liquidi noti che coprono le tre famiglie principali—a bastoncino, discotici e a nucleo piegato—mentre il resto sono composti non cristalli liquidi presi da vari ambiti della chimica. Con questa raccolta hanno addestrato e confrontato diversi modelli di machine learning per distinguere le molecole a cristallo liquido da tutte le altre. Combinando tre modelli complementari in uno schema a “voto di maggioranza” che privilegia l’individuazione di un candidato come cristallo liquido piuttosto che perderlo, il classificatore ha recuperato correttamente quasi tutti i cristalli liquidi noti in un test indipendente, con prestazioni particolarmente solide per i tipi più complessi discotici e a nucleo piegato.
Prevedere le temperature chiave dalla forma molecolare
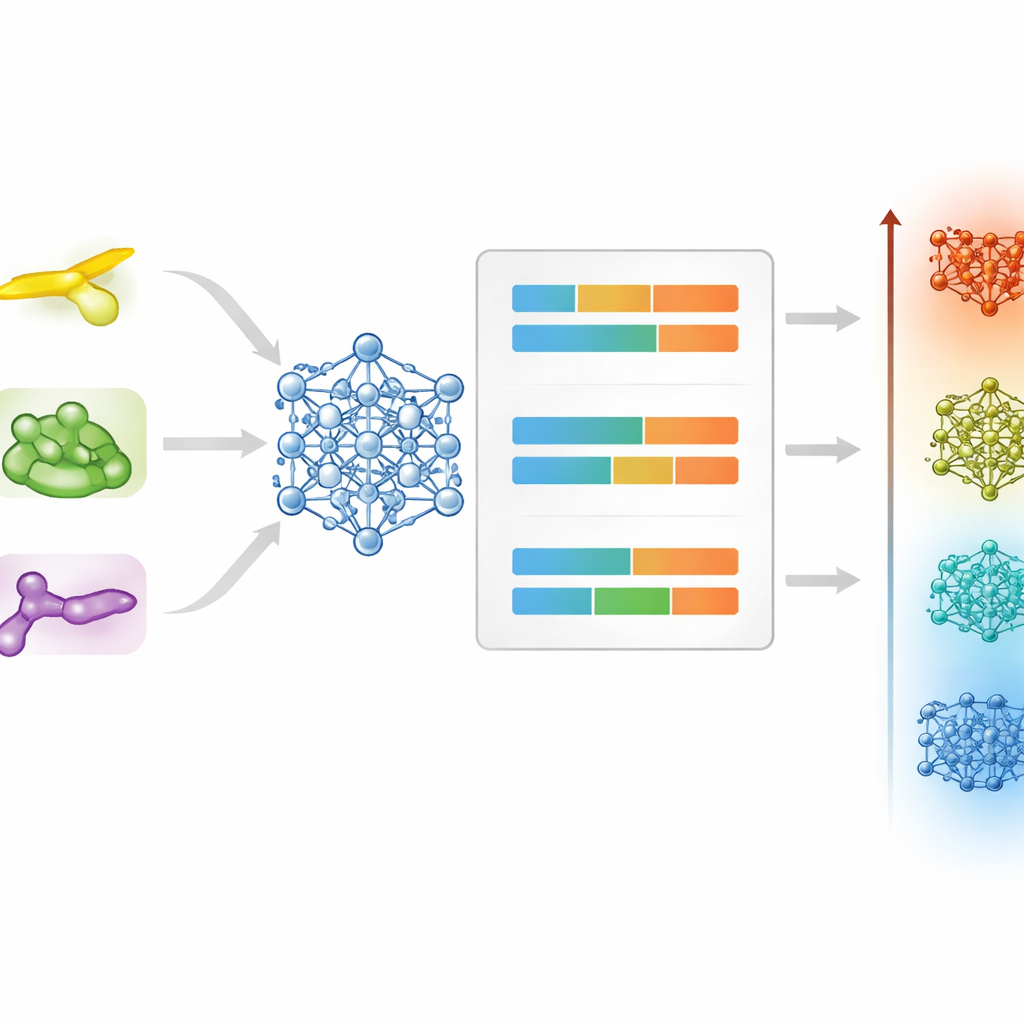
Una volta che una molecola è stata etichettata come probabile cristallo liquido, la sfida successiva è prevederne le temperature di fusione e di chiarimento. Per affrontare questo problema, il team ha confrontato algoritmi tradizionali che si basano su fingerprint pre‑calcolati della struttura molecolare con un approccio più recente che tratta ogni molecola come un grafo di atomi e legami. Per le temperature di fusione, un modello ibrido che combina un regressore random‑forest con una rete neurale grafica ha fornito la migliore accuratezza complessiva, gestendo con successo i comportamenti differenti di materiali a bastoncino, discotici e a nucleo piegato. Per le temperature di chiarimento, il modello basato su grafi da solo ha generalizzato meglio, probabilmente perché questa transizione a temperature più elevate dipende maggiormente dalla forma globale e dalla connettività della molecola piuttosto che da dettagli locali.
Individuare pattern sottili e comprendere gli errori
Una prova severa per qualsiasi strumento predittivo è se riesce a cogliere i pattern delicati che i chimici conoscono per esperienza. Qui, i modelli non solo hanno riprodotto i tipici intervalli di temperatura ma hanno anche rispecchiato i cosiddetti effetti dispari‑pari, dove l’aggiunta o la rimozione di un singolo carbonio in una catena laterale causa un’oscillazione nelle temperature di transizione. Gli autori hanno inoltre esaminato i casi in cui le previsioni erano errate di oltre circa 30 gradi. Queste molecole problematiche spesso presentavano scheletri molto curvi, nuclei voluminosi a forma di disco o sostituenti insoliti come multiple atomi di fluoro, tutti elementi che possono modificare il modo di impacchettamento molecolare in modi che i dati e i descrittori attuali non catturano completamente. Questa analisi indica dove ulteriori esperimenti e descrittori più raffinati potrebbero migliorare lo strumento.
Una nuova scorciatoia per progettare i materiali del futuro
Tutti i dati, i modelli e un’interfaccia web user‑friendly sono resi disponibili apertamente come Liquid Crystal Predictor. Un ricercatore può ora disegnare o specificare una molecola, far decidere alla piattaforma se è probabile che formi una fase a cristallo liquido e stimare le temperature alle quali essa apparirà e scomparirà—senza necessitare di una profonda competenza in machine learning. Sebbene l’accuratezza sia ancora più bassa per alcune famiglie molecolari meno rappresentate, il sistema offre già una guida potente per lo screening dei candidati prima della sintesi e dei test. Col tempo, man mano che verranno aggiunte strutture più esotiche e descrittori strutturali migliori, strumenti come questo potrebbero trasformare la ricerca di materiali avanzati per cristalli liquidi da un’arte in una scienza collaborativa guidata dai dati.
Citazione: Wu, H., Patel, H., Xiang, Y. et al. Liquid crystal predictor: a machine learning platform for classification and phase transition forecast. npj Soft Matter 2, 11 (2026). https://doi.org/10.1038/s44431-026-00020-1
Parole chiave: cristalli liquidi, machine learning, transizioni di fase, progettazione dei materiali, materia soffice