Clear Sky Science · sv
CDK4/6‑hämmare återställer PARP‑hämmarresistens genom att rikta in sig på E2F1–MCM2/5‑vägen
Varför denna cancerforskning är viktig
Äggstockscancer är en av de dödligaste cancerformerna som drabbar personer med äggstockar, delvis eftersom den ofta återkommer efter behandling. En läkemedelsklass kallad PARP‑hämmare har gett nytt hopp genom att selektivt angripa tumörceller med brister i DNA‑reparation. Men många tumörer hittar så småningom sätt att undkomma dessa läkemedel och börjar växa igen. Denna studie avslöjar hur vissa äggstockscancer utvecklar resistens mot en vanligt använd PARP‑hämmare och visar att tillsats av en annan typ av läkemedel kan slå av den resistensen och krympa tumörer igen.
När ett genombrottsläkemedel slutar fungera
PARP‑hämmare, inklusive läkemedlet niraparib, fungerar genom att överbelasta cancerceller som redan är dåliga på att laga brutet DNA. Inledningsvis svarar många äggstockstumörer väl, men de flesta patienter drabbas så småningom av återfall när cancern anpassar sig. Forskarna skapade i labbet cellinjer av äggstockscancer som var resistenta mot niraparib genom att utsätta cellerna för läkemedlet under många månader. Jämfört med de ursprungliga, läkemedelskänsliga cellerna fortsatte de resistenta cellerna att dela sig, bildade fler kolonier och stoppade inte längre sin cellcykel när de exponerades för niraparib. Dessa modeller gjorde det möjligt för teamet att ställa en central fråga: vilka förändringar inne i cellerna gör att de kan ignorera en behandling som tidigare stoppade dem?
DNA‑kopieringsmaskinerna skruvar upp volymen
Genom att läsa av vilka gener som var på‑ eller avstängda i både känsliga och resistenta celler fann teamet ett slående mönster. Två medlemmar i en proteinfamilj som hjälper till att kopiera DNA—kallade MCM2 och MCM5—sänktes av niraparib i normala celler, men var starkt uppreglerade i resistenta celler. Proteinerna bildade ett tätare partnerskap i de resistenta cellerna, och kliniska data visade att äggstockstumörer med högre nivåer av dessa två proteiner var kopplade till sämre patientutfall. När forskarna avsiktligt sänkte MCM2 eller MCM5 i resistenta celler blev dessa celler återigen sårbara för niraparib, visade mer DNA‑skada och delade sig långsammare. Omvänt gjorde tvångsmässig överproduktion av MCM2 eller MCM5 cellerna svårare att döda med PARP‑hämmare, vilket stöder idén att detta DNA‑kopieringsduo är en nyckeldrivkraft bakom resistensen.
En kontrollbrytare som matar på resistensen
Forskarna frågade sig sedan vad som styr ökningen av MCM2 och MCM5 i resistenta celler. De fokuserade på ett protein kallat E2F1, en huvudomkopplare som aktiverar många gener som behövs för att celler ska kopiera sitt DNA. Med hjälp av offentliga cancerdata, förutsägelser om DNA‑bindning och biokemiska tester visade de att E2F1 direkt fäster vid på‑slagsregionerna för MCM2‑ och MCM5‑generna och ökar deras aktivitet. Ökad E2F1 i känsliga celler höjde MCM2/5‑nivåerna och gjorde cellerna mer läkemedelsresistenta, medan dämpning av E2F1 i resistenta celler sänkte MCM2/5 och återställde känsligheten för niraparib. I dessa resistenta celler var också ett par proteiner kallade CDK4 och CDK6—välkända drivkrafter för celldelning—uppreglerade, vilket tyder på en händelsekedja från CDK4/6 via E2F1 till MCM2/5 som skyddar tumörcellerna från PARP‑hämmare.
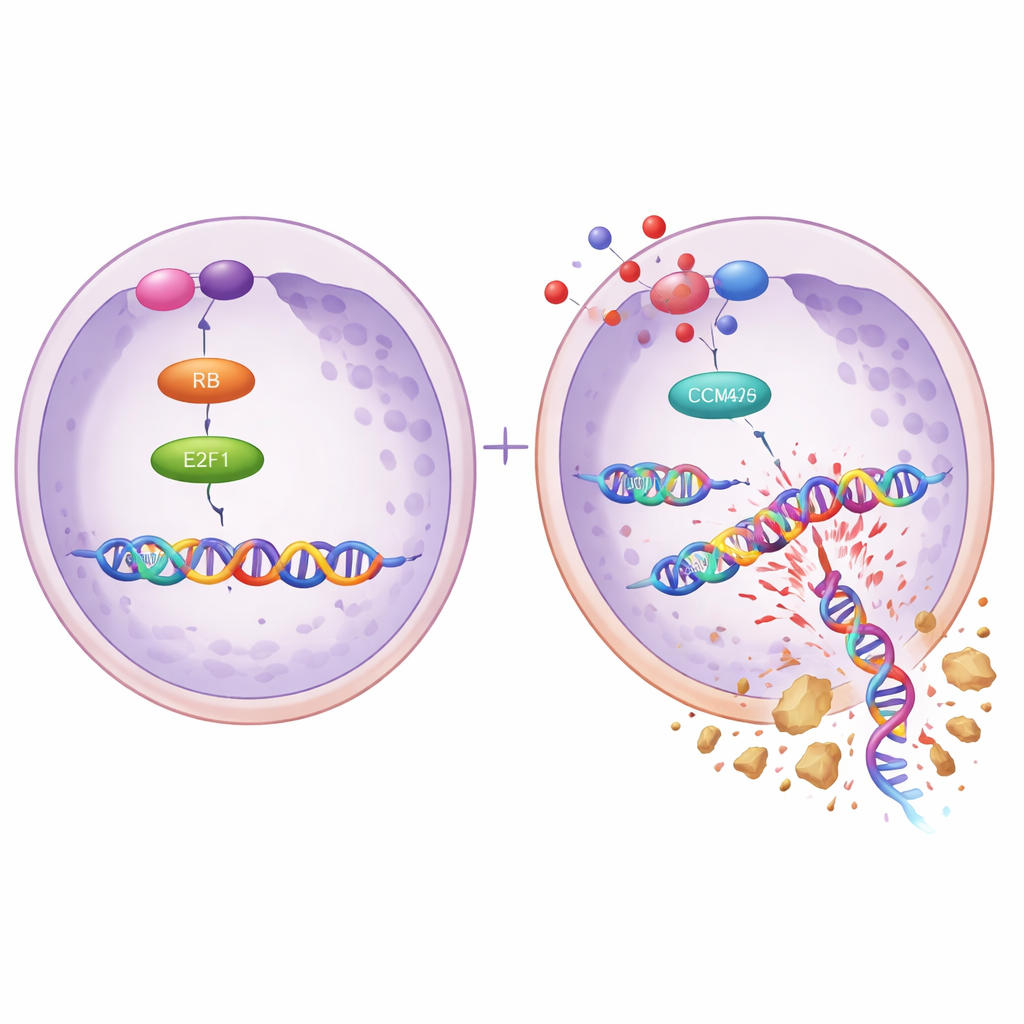
Att använda ett läkemedel för att få ett annat att fungera igen
Eftersom CDK4/6‑proteiner sitter uppströms om E2F1 testade teamet om blockering av CDK4/6 kunde slå av hela resistensprogrammet. De kombinerade niraparib med dalpiciclib, en CDK4/6‑hämmare som redan används kliniskt för andra cancerformer, och fann att de två läkemedlen fungerade bättre tillsammans än var och en för sig i resistenta äggstockscellinjer. Kombinationen minskade celltillväxt, kolonibildning och DNA‑kopieringsaktivitet, och försvagade partnerskapet mellan MCM2 och MCM5. Markörer för DNA‑skada ökade kraftigt, medan reparationsrelaterade proteiner sjönk. Liknande effekter sågs med en annan CDK4/6‑hämmare, abemaciclib. Hos möss med niraparib‑resistenta äggstockstumörer krympte tumörerna mycket mer med kombinationen dalpiciclib och niraparib än med något av läkemedlen ensamt, och tumörprover visade lägre nivåer av MCM2, MCM5 och E2F1.
Vad detta betyder för framtida behandling av äggstockscancer
Enkelt uttryckt visar studien att vissa äggstockscancer övervinner PARP‑hämmare genom att skruva upp sin DNA‑kopieringsmaskin, ledd av MCM2 och MCM5 under kontroll av E2F1‑omkopplaren och dess CDK4/6‑regulatorer. Genom att lägga till en CDK4/6‑hämmare kan läkare kanske sänka denna maskin igen, blotta tumörens svaga punkt i DNA‑reparation och göra PARP‑hämmare effektiva på nytt. Även om kliniska prövningar krävs, lägger arbetet fram en tydlig biologisk karta för att kombinera dessa läkemedel och framhäver MCM2/5 som lovande markörer—och potentiella mål—för att övervinna behandlingsresistens vid äggstockscancer.
Citering: Feng, Y., Fu, M., Zheng, B. et al. CDK4/6i reverse PARPi resistance by targeting the E2F1- MCM2/5 pathway. npj Precis. Onc. 10, 162 (2026). https://doi.org/10.1038/s41698-026-01353-w
Nyckelord: äggstockscancer, resistens mot PARP‑hämmare, CDK4/6‑hämmare, DNA‑replikation, kombinationer av riktade behandlingar