Clear Sky Science · de
CDK4/6i kippt PARPi-Resistenz, indem der E2F1–MCM2/5-Weg angegriffen wird
Warum diese Krebsforschung wichtig ist
Das Ovarialkarzinom gehört zu den tödlichsten Krebsarten bei Personen mit Eierstöcken, teilweise weil es nach einer Behandlung häufig wieder auftritt. Eine Wirkstoffklasse namens PARP-Inhibitoren hat neue Hoffnungen geweckt, da sie gezielt Tumorzellen mit defekter DNA-Reparatur angreift. Dennoch finden viele Tumoren schließlich Wege, diesen Medikamenten zu entkommen, und wachsen wieder. Diese Studie zeigt, wie einige Ovarialkarzinome Resistenz gegen einen weit verbreiteten PARP-Inhibitor entwickeln, und demonstriert, dass das Hinzufügen eines weiteren Wirkstoffs diese Resistenz ausschalten und die Tumoren erneut verkleinern kann.
Wenn ein Durchbruchsmedikament nicht mehr wirkt
PARP-Inhibitoren, darunter das Medikament Niraparib, wirken, indem sie Krebszellen überfordern, die bereits schlecht darin sind, gebrochene DNA zu reparieren. Zunächst sprechen viele Ovarialtumoren gut an, doch die meisten Patientinnen und Patienten erleben letztlich ein Rezidiv, weil sich der Krebs anpasst. Die Forschenden erzeugten im Labor Niraparib‑resistente Ovarialkrebszelllinien, indem sie die Zellen über mehrere Monate dem Medikament aussetzten. Im Vergleich zu den ursprünglichen, medikamentensensitiven Zellen teilten sich die resistenten Zellen weiterhin, bildeten mehr Kolonien und blieben beim Kontakt mit Niraparib nicht länger im Zellzyklus stehen. Diese Modelle ermöglichten dem Team, eine zentrale Frage zu stellen: Welche Veränderungen in den Zellen erlauben es ihnen, eine Behandlung abzuschütteln, die sie einst stoppte?
Die DNA-Kopiermaschine wird lauter gestellt
Durch das Ablesen, welche Gene in sensiblen und resistenten Zellen an- oder abgeschaltet waren, fand das Team ein auffälliges Muster. Zwei Mitglieder einer Proteinfamilie, die beim Kopieren der DNA helfen — MCM2 und MCM5 — wurden in normalen Zellen durch Niraparib herunterreguliert, in resistenten Zellen hingegen stark hochreguliert. Die Proteine arbeiteten in resistenten Zellen enger zusammen, und klinische Daten zeigten, dass Ovarialtumoren mit höheren Mengen dieser beiden Proteine mit schlechteren Patientenergebnissen einhergingen. Wenn die Forschenden MCM2 oder MCM5 in resistenten Zellen gezielt reduzierten, wurden diese Zellen wieder empfindlich gegenüber Niraparib, zeigten mehr DNA-Schäden und teilten sich langsamer. Umgekehrt machte die erzwungene Überexpression von MCM2 oder MCM5 Zellen schwerer mit PARP-Inhibitoren zu töten, was die Idee stützt, dass dieses DNA-Kopierduo ein Schlüsseltreiber der Resistenz ist.
Ein Steuerungsschalter, der die Resistenz antreibt
Die Forschenden fragten anschließend, was den Anstieg von MCM2 und MCM5 in resistenten Zellen steuert. Sie konzentrierten sich auf ein Protein namens E2F1, einen Master‑Schalter, der viele Gene für die DNA-Replikation einschaltet. Mit Hilfe öffentlicher Krebsdaten, Vorhersagen zu DNA‑Bindungsstellen und biochemischen Tests zeigten sie, dass E2F1 direkt an die Aktivatorregionen der MCM2‑ und MCM5‑Gene bindet und deren Aktivität steigert. Eine Erhöhung von E2F1 in sensiblen Zellen hob die MCM2/5‑Spiegel an und machte die Zellen resistenter gegen das Medikament, während das Abschwächen von E2F1 in resistenten Zellen MCM2/5 senkte und die Niraparib‑Empfindlichkeit wiederherstellte. In diesen resistenten Zellen waren außerdem die beiden Proteine CDK4 und CDK6 — bekannte Treiber der Zellteilung — erhöht, was eine Ereigniskette von CDK4/6 über E2F1 zu MCM2/5 nahelegt, die Tumorzellen vor PARP-Inhibitoren schützt.
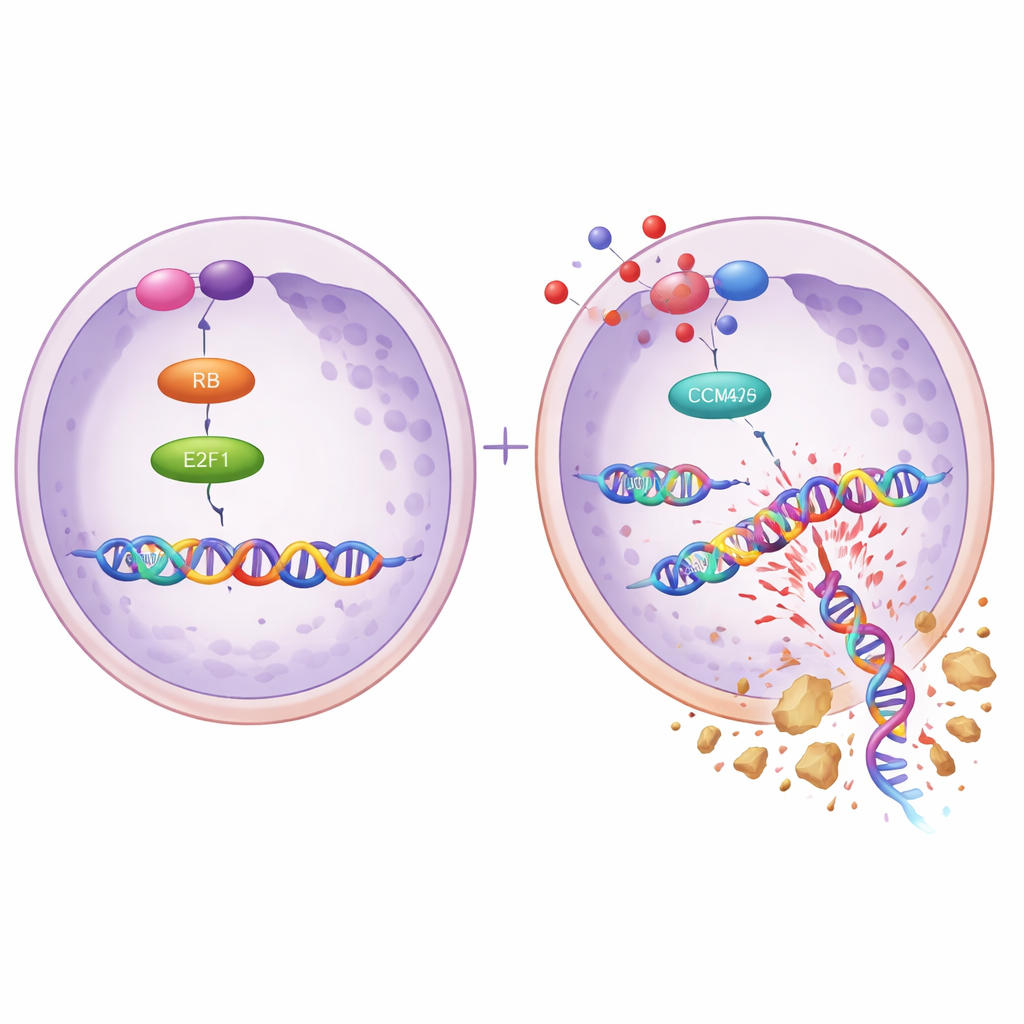
Ein Medikament einsetzen, damit ein anderes wieder wirkt
Da CDK4/6 oberhalb von E2F1 angesiedelt sind, testete das Team, ob die Blockade von CDK4/6 das gesamte Resistenzprogramm wieder abschalten kann. Sie kombinierten Niraparib mit Dalpiciclib, einem CDK4/6‑Inhibitor, der bereits klinisch für andere Krebsarten eingesetzt wird, und fanden, dass die beiden Wirkstoffe in resistenten Ovarialkrebszellen zusammen besser wirkten als jeder für sich. Die Kombination verringerte Zellwachstum, Koloniebildung und DNA‑Replikationsaktivität und schwächte die Partnerschaft zwischen MCM2 und MCM5. Marker für DNA‑Schäden stiegen deutlich an, während Proteine, die mit der Reparatur zu tun haben, zurückgingen. Ähnliche Effekte wurden mit einem anderen CDK4/6‑Inhibitor, Abemaciclib, beobachtet. Bei Mäusen mit Niraparib‑resistenten Ovarialtumoren schrumpften die Tumoren unter der Kombination von Dalpiciclib und Niraparib deutlich stärker als bei einer Einzelbehandlung, und Tumorproben zeigten niedrigere Mengen an MCM2, MCM5 und E2F1.
Was das für die zukünftige Behandlung von Ovarialkarzinom bedeutet
Vereinfacht gesagt zeigt die Studie, dass einige Ovarialkarzinome PARP-Inhibitoren dadurch überwinden, dass sie ihre DNA‑Kopiermaschinen hochfahren — angeführt von MCM2 und MCM5 unter Kontrolle des E2F1‑Schalters und dessen CDK4/6‑Regulatoren. Durch das Hinzufügen eines CDK4/6‑Inhibitors könnten Ärztinnen und Ärzte diese Maschinerie wieder herunterregeln, die Verwundbarkeit des Tumors in der DNA‑Reparatur erneut freilegen und PARP-Inhibitoren wieder wirksam machen. Zwar sind klinische Studien erforderlich, doch die Arbeit liefert eine klare biologische Roadmap für die Kombination dieser Medikamente und hebt MCM2/5 als vielversprechende Marker — und potenzielle Ziele — hervor, um Therapieresistenz beim Ovarialkarzinom zu überwinden.
Zitation: Feng, Y., Fu, M., Zheng, B. et al. CDK4/6i reverse PARPi resistance by targeting the E2F1- MCM2/5 pathway. npj Precis. Onc. 10, 162 (2026). https://doi.org/10.1038/s41698-026-01353-w
Schlüsselwörter: Ovarialkarzinom, Resistenz gegen PARP-Inhibitoren, CDK4/6-Inhibitoren, DNA-Replikation, kombinierte gezielte Therapien