Clear Sky Science · pt
Mapeando o cenário de inibição da P-glicoproteína via docking em conjunto conformacional
Por que essa bomba celular importa para a medicina
Muitos medicamentos modernos falham não porque não atinjam seu alvo, mas porque nossas próprias células os expulsam. Um dos principais culpados é a P-glicoproteína, uma pequena bomba embutida nas membranas celulares que expele uma ampla variedade de fármacos, desde quimioterápicos até antidepressivos. Este estudo faz uma pergunta simples, mas crucial: podemos desenhar bloqueadores melhores dessa bomba olhando não para uma estrutura congelada única, mas para as várias formas que ela assume enquanto se move e funciona? A resposta ajuda a explicar por que inibidores anteriores frequentemente decepcionaram e aponta caminhos mais inteligentes para domar essa bomba no futuro.
Um porteiro mutável nas paredes celulares
A P-glicoproteína fica em barreiras protetoras como a parede intestinal e a barreira hemato‑encefálica, onde atua como um seguranças, expulsando moléculas estranhas de volta para a corrente sanguínea. Ela faz isso alternando entre várias conformações que se abrem para o interior da célula, se fecham e então se abrem para fora para expelir substâncias. Projetos de fármacos anteriores frequentemente trataram essa bomba como se tivesse uma única forma dominante. Na realidade, ela é flexível e espaçosa, com vários bolsões sobrepostos que acomodam muitos tipos de químicos. Essa flexibilidade a torna muito eficiente em expulsar drogas, mas também dificulta bloqueá‑la de maneira precisa e segura.
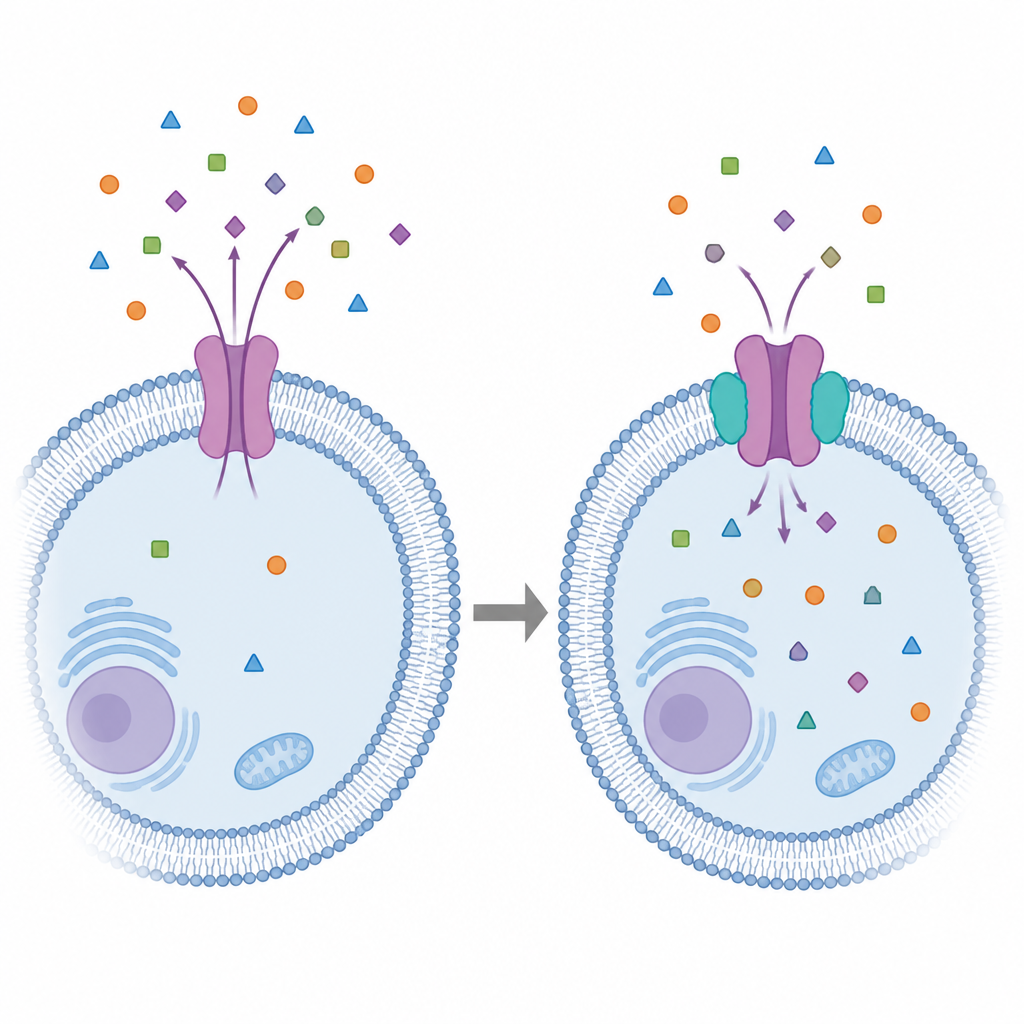
Simulando um alvo em movimento
Para capturar esse movimento, os pesquisadores utilizaram longas simulações computacionais para observar a P-glicoproteína humana inserida em uma membrana realista composta de lipídios e colesterol. Eles partiram de estruturas detalhadas obtidas por crio‑microscopia eletrônica e de modelos gerados por inteligência artificial, e então deixaram a proteína se movimentar por centenas de nanosegundos. A partir desses “filmes” extraíram 22 conformações representativas, que vão de estados mais abertos voltados para o interior até estados mais fechados e ocluídos, e também incluíram um linker flexível previamente não resolvido que conecta partes da proteína. Isso criou um “conjunto” estrutural diverso que reflete melhor como a bomba realmente se comporta na membrana celular.
Testando dezenas de inibidores em muitas formas
Em seguida, a equipe encaixou (docked) 60 bloqueadores conhecidos da P‑glicoproteína em cada uma das 22 conformações, avaliando quão firmemente cada composto poderia se ligar. Em vez de um quadro uniforme, observaram preferências claras. Mais da metade dos inibidores favoreceu uma conformação voltada para o interior que apresentava tanto um inibidor ligado quanto o linker flexível presente, sugerindo que essa combinação cria uma cavidade justa e bem formada. Em contraste, um grupo de fármacos grandes e em anel, como ciclosporina e valsopodar, se saiu melhor em modelos onde um portal lateral formado por duas hélices estava mais amplo, fornecendo espaço extra para acomodar seu tamanho volumoso. Esses padrões foram reforçados por cálculos energéticos mais detalhados em pares selecionados de droga e bomba.
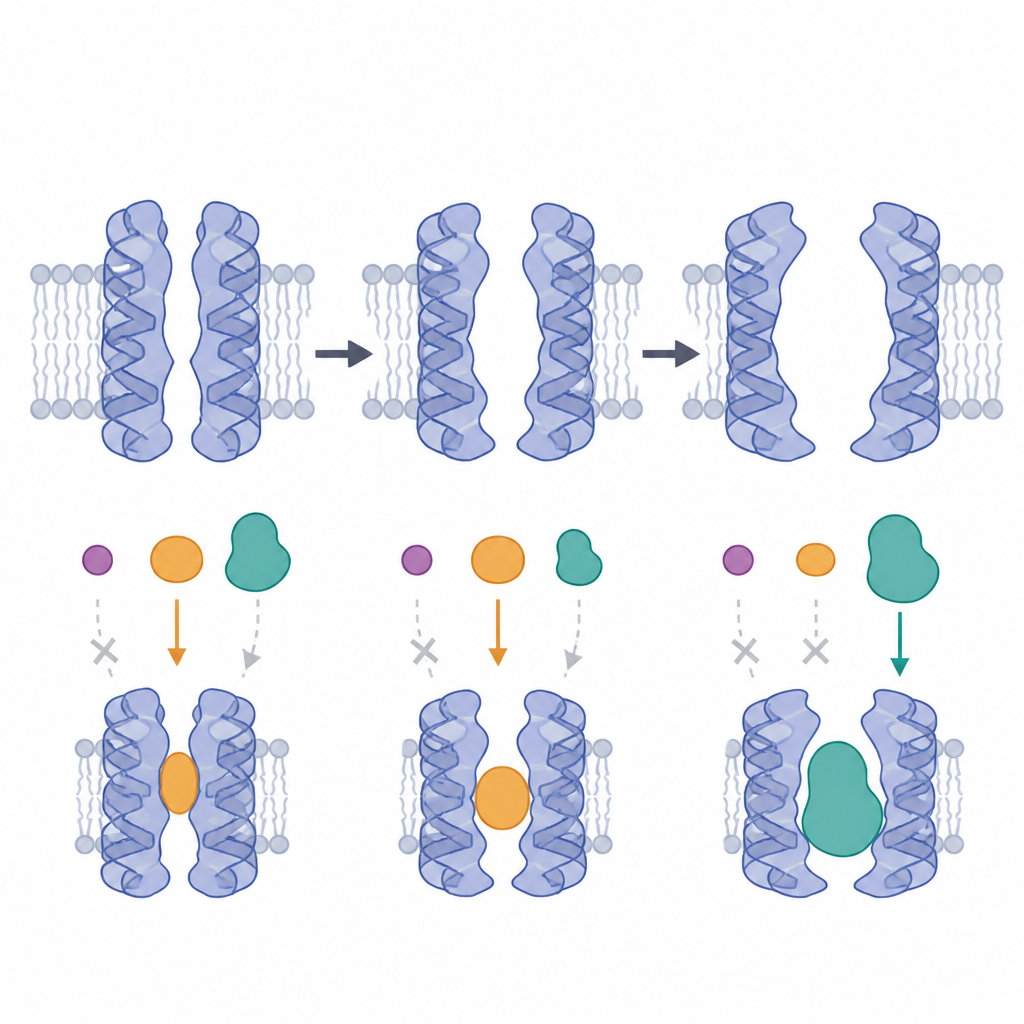
Onde os fármacos tendem a se ancorar na bomba
Ao examinar onde cada composto se acomodou dentro da bomba, os autores mapearam três bolsões principais. Um desses bolsões, envolvendo várias hélices-chave, emergiu como o sítio de ligação mais usado, especialmente na conformação favorecida que continha inibidor e linker. Certos aminoácidos nessa região apareciam repetidamente em contato com muitos inibidores, correspondendo a indícios experimentais anteriores sobre pontos críticos de interação. Ao mesmo tempo, fármacos quimicamente semelhantes nem sempre preferiam a mesma conformação ou bolsão, ressaltando que pequenos ajustes em tamanho e flexibilidade podem deslocar qual conformação lhes convém melhor. Isso significa que projetar bloqueadores eficazes requer pensar tanto na arquitetura da droga quanto no cenário móvel da bomba.
O que isso significa para o desenho de fármacos no futuro
Para um público não especializado, a mensagem chave é que a P‑glicoproteína não é uma fechadura rígida, mas um portão em constante mudança, e bloqueadores bem‑sucedidos precisam ser casados a formas específicas desse portão. O estudo mostra que certas conformações, especialmente as que incluem o linker flexível, parecem particularmente atraentes para muitos inibidores, enquanto drogas macrocilíndricas volumosas precisam de um portal mais aberto. Ao combinar o movimento realista da proteína com o docking sistemático de muitos compostos, o trabalho traça um roteiro para projetar bloqueadores de bomba mais seletivos e potencialmente mais seguros. Em vez de buscar um único inibidor perfeito, esforços futuros podem se concentrar em direcionar estados particulares da bomba que melhor se ajustem a uma dada classe de drogas, com o objetivo de longo prazo de ajudar medicamentos importantes a permanecerem dentro das células tempo suficiente para fazer seu trabalho.
Citação: Elbahnsi, A., Dragomirescu, C.D., Palumbo, N. et al. Mapping the inhibition landscape of P-glycoprotein via conformational ensemble docking. Sci Rep 16, 15393 (2026). https://doi.org/10.1038/s41598-026-46760-y
Palavras-chave: P-glicoproteína, resistência a múltiplos fármacos, bomba de efluxo de drogas, docking molecular, transportador ABC