Clear Sky Science · de
Kartierung der Hemmungslandschaft von P‑Glykoprotein mittels konformationellem Ensemble‑Docking
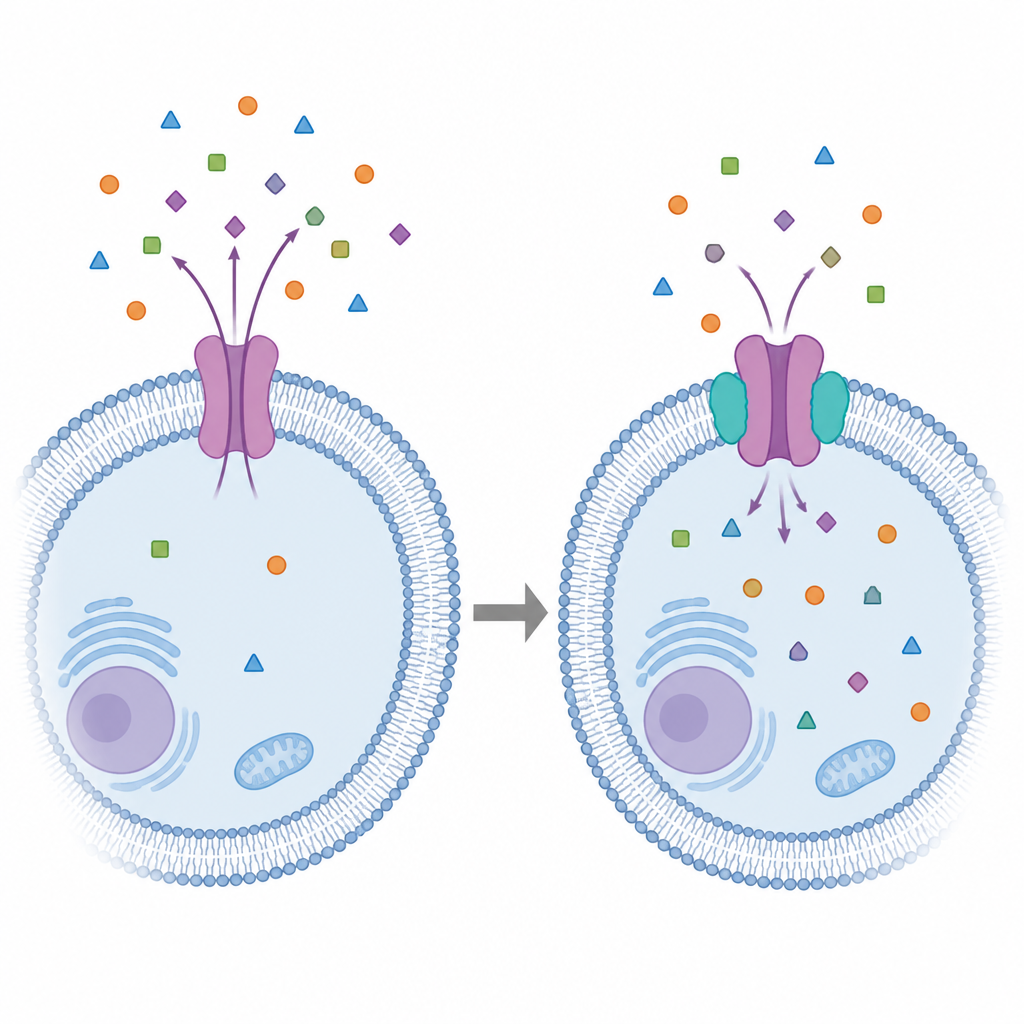
Warum diese Zellpumpe für die Medizin wichtig ist
Viele moderne Medikamente scheitern nicht, weil sie ihr Ziel verfehlen, sondern weil unsere eigenen Zellen sie wieder hinauspumpen. Ein zentraler Übeltäter ist das P‑Glykoprotein, eine winzige Pumpe in Zellmembranen, die ein breites Spektrum an Arzneistoffen hinausbefördert – von Krebsmedikamenten bis zu Antidepressiva. Diese Studie stellt eine einfache, aber entscheidende Frage: Können wir bessere Hemmstoffe dieser Pumpe entwickeln, wenn wir nicht von einer einzigen starren Struktur ausgehen, sondern die vielen Formen betrachten, die sie im Betrieb annimmt? Die Antwort hilft zu erklären, warum frühere Inhibitoren oft enttäuschten, und weist auf klügere Wege hin, diese Pumpe künftig zu zähmen.
Ein formwandelnder Wächter in Zellwänden
P‑Glykoprotein sitzt in Schutzbarrieren wie der Darmwand und der Blut‑Hirn‑Schranke und wirkt dort wie ein Türsteher, der Fremdstoffe zurück in den Blutkreislauf befördert. Es tut dies, indem es mehrere Konformationen durchläuft: nach innen geöffnete Zustände, Verschluss und anschließend eine nach außen geöffnete Form, um Substanzen auszustoßen. Frühere Wirkstoffentwicklung behandelte diese Pumpe häufig so, als habe sie nur eine dominante Form. In Wirklichkeit ist sie flexibel und geräumig, mit mehreren überlappenden Taschen, die viele verschiedene Chemikalien aufnehmen können. Diese Flexibilität macht sie effizient beim Ausstoß von Medikamenten, erschwert aber zugleich, die Pumpe selbst gezielt und sicher zu blockieren.
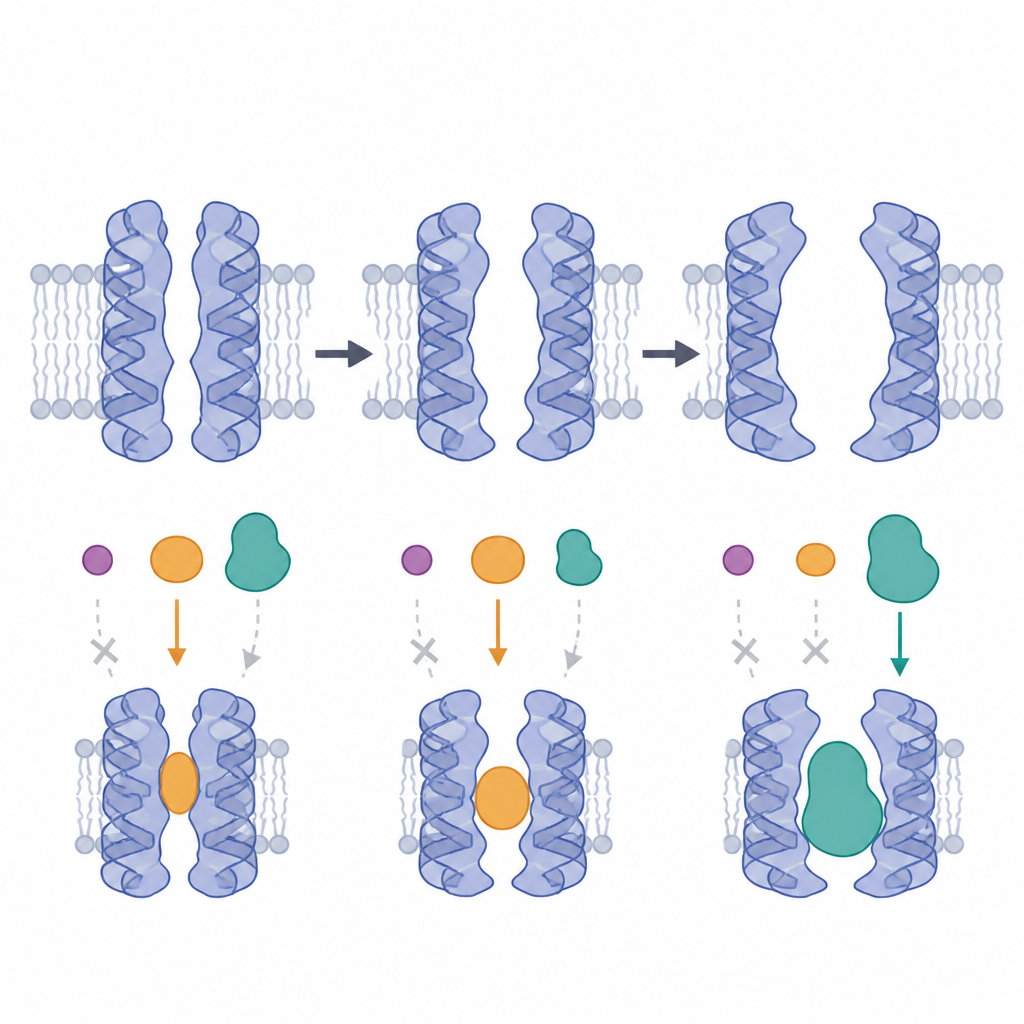
Ein bewegliches Ziel simulieren
Um diese Bewegung einzufangen, verwendeten die Forschenden lange Computersimulationen, um das menschliche P‑Glykoprotein in einer realistischen Membran aus Lipiden und Cholesterin zu beobachten. Sie starteten von detaillierten Strukturen aus Kryo‑Elektronenmikroskopie und von Modellen künstlicher Intelligenz und ließen das Protein hunderte Nanosekunden bewegen. Aus diesen «Filmen» extrahierten sie 22 repräsentative Formen, die von offeneren, nach innen gerichteten Zuständen bis zu geschlosseneren, okkludierten Varianten reichten, und integrierten zudem einen zuvor unaufgelösten flexiblen Linker, der Teile des Proteins verbindet. So entstand ein vielfältiges strukturelles Ensemble, das besser widerspiegelt, wie die Pumpe tatsächlich in der Membran agiert.
Dutzende Inhibitoren in vielen Formen getestet
Das Team dockte anschließend 60 bekannte P‑Glykoprotein‑Blocker in jede der 22 Formen und bewertete, wie stark jede Verbindung binden könnte. Statt eines einheitlichen Bildes zeigten sich deutliche Präferenzen. Mehr als die Hälfte der Inhibitoren bevorzugte eine bestimmte nach innen gerichtete Konformation, in der sowohl ein gebundener Inhibitor als auch der flexible Linker vorhanden waren, was darauf hindeutet, dass diese Kombination eine eng anliegende, wohlgeformte Höhlung erzeugt. Im Gegensatz dazu schnitt eine Gruppe großer, ringförmiger Wirkstoffe, wie Cyclosporin und Valspodar, in Modellen besser ab, in denen ein Seitenportal aus zwei Helices weiter geöffnet war und so zusätzlichen Platz für ihre sperrige Gestalt bot. Diese Muster wurden durch detailliertere Energie‑Berechnungen an ausgewählten Wirkstoff‑Paaren gestützt.
Wo Medikamente typischerweise an der Pumpe anhaften
Indem die Autoren untersuchten, wo sich jede Verbindung innerhalb der Pumpe niederließ, kartierten sie drei Haupttaschen. Eine Tasche, an der mehrere Schlüsselhelix‑Abschnitte beteiligt sind, erwies sich als am häufigsten genutzte Bindungsstelle, besonders in der bevorzugten Inhibitor‑gebundenen, Linker‑enthaltenden Form. Bestimmte Aminosäuren in diesem Bereich traten wiederholt in Kontakt mit vielen Inhibitoren auf und stimmten mit früheren experimentellen Hinweisen auf entscheidende Interaktions‑Hotspots überein. Gleichzeitig bevorzugten chemisch ähnliche Medikamente nicht immer dieselbe Pumpenform oder Tasche, was unterstreicht, dass kleine Änderungen in Größe und Flexibilität verschieben können, welche Konformation am besten passt. Das bedeutet: Effektives Design von Hemmstoffen muss sowohl die Architektur des Wirkstoffs als auch die bewegliche Landschaft der Pumpe berücksichtigen.
Was das für zukünftiges Wirkstoffdesign bedeutet
Für Nicht‑Spezialisten ist die Kernbotschaft: P‑Glykoprotein ist kein starrer Schließmechanismus, sondern ein ständig wechselndes Tor, und erfolgreiche Hemmstoffe müssen auf bestimmte Formen dieses Tors abgestimmt werden. Die Studie zeigt, dass gewisse Konformationen, insbesondere solche mit dem flexiblen Linker, für viele Inhibitoren besonders attraktiv erscheinen, während sperrige makrocyclische Wirkstoffe ein weiter geöffnetes Portal benötigen. Durch die Kombination realistischer Proteinbewegung mit systematischem Docking vieler Verbindungen skizziert die Arbeit einen Fahrplan für die Entwicklung selektiverer und potenziell sichererer Pumpenhemmer. Statt nach einem einzelnen perfekten Inhibitor zu suchen, könnten künftige Ansätze darauf abzielen, spezifische Zustände der Pumpe ins Visier zu nehmen, die am besten zu einer bestimmten Wirkstoffklasse passen, mit dem langfristigen Ziel, wichtigen Medikamenten ausreichend Verweildauer in Zellen zu ermöglichen, damit sie ihre Wirkung entfalten können.
Zitation: Elbahnsi, A., Dragomirescu, C.D., Palumbo, N. et al. Mapping the inhibition landscape of P-glycoprotein via conformational ensemble docking. Sci Rep 16, 15393 (2026). https://doi.org/10.1038/s41598-026-46760-y
Schlüsselwörter: P‑Glykoprotein, multiresistente, Wirkstoff‑Effluxpumpen, molekulares Docking, ABC‑Transporter