Clear Sky Science · pt
Engenharia de características multi-ômicas guiada por modelos de base biomédicos melhora a predição de resposta a medicamentos para pacientes com doença inflamatória intestinal
Por que esta pesquisa é importante para os pacientes
Pessoas com doença inflamatória intestinal (DII) frequentemente enfrentam um processo frustrante de tentativa e erro ao iniciar novos medicamentos. O mesmo fármaco pode acalmar uma inflamação intestinal severa em uma pessoa e mal ajudar outra. Este estudo investiga como poderosos modelos de inteligência artificial (IA), originalmente desenvolvidos para entender a biologia em larga escala, podem ser reaproveitados para extrair muito mais informação de conjuntos de dados pequenos de pacientes. O objetivo é simples, porém ambicioso: usar o perfil genético e molecular de cada pessoa para prever antecipadamente quão bem ela responderá a um medicamento comum para DII.
Usando IA de grande porte para ajudar estudos pequenos
Ferramentas tradicionais de IA médica têm dificuldade quando o número de pacientes é pequeno, mesmo que cada paciente seja profundamente caracterizado. Neste trabalho, os pesquisadores inverteram esse problema ao emprestar conhecimento de um enorme “modelo de base” chamado MAMMAL, treinado originalmente em vastas coleções de dados de proteínas e fármacos. Em vez de retreinar esse modelo, eles o usam como uma calculadora inteligente para estimar quão fortemente um dado fármaco se liga a milhares de proteínas humanas. Essas pontuações de força de ligação tornam-se então novas características ricas em informação que podem ser inseridas em modelos de aprendizado de máquina mais modestos, adaptados aos 51 pacientes com DII deste estudo.
Observando o intestino: DNA e RNA
A equipe trabalhou com amostras de tecido coletadas dos intestinos inflamados de pessoas com colite ulcerativa ou doença de Crohn. De cada amostra, mediram duas camadas principais da biologia: DNA, focando em pequenas alterações genéticas chamadas SNPs, e RNA, que reflete o nível de atividade de cada gene no tecido. Também expuseram o tecido à prednisolona, um tratamento esteroide de primeira linha para DII, e mediram quanto um sinal inflamatório-chave chamado TNFα reduzia em resposta. Essa mudança no TNFα serviu como um substituto de quão bem o tecido do paciente respondeu ao medicamento. O desafio foi conectar milhares de medidas genéticas e moleculares a esse desfecho de resposta ao medicamento.
Ensinando aos modelos onde prestar atenção
O primeiro passo foi decidir quais proteínas e genes os modelos deveriam priorizar. Usando o modelo de base, os pesquisadores estimaram o quão firmemente a prednisolona se ligaria a mais de 11.000 proteínas humanas, e então focaram nos 5% superiores com maior ligação prevista. De forma encorajadora, essa lista curta incluiu alvos conhecidos do fármaco e proteínas envolvidas em detecção química e sinalização, sugerindo que as classificações da IA faziam sentido biologicamente. Eles então combinaram essa lista com as alterações no DNA de cada paciente, criando sequências de proteínas “mutadas” personalizadas e recalculando como cada mutação poderia, ligeiramente, fortalecer ou enfraquecer a ligação da prednisolona. Essas mudanças previstas na força de ligação tornaram-se características compactas e específicas por paciente, capturando como variantes em proteínas-chave poderiam alterar a ação do medicamento.
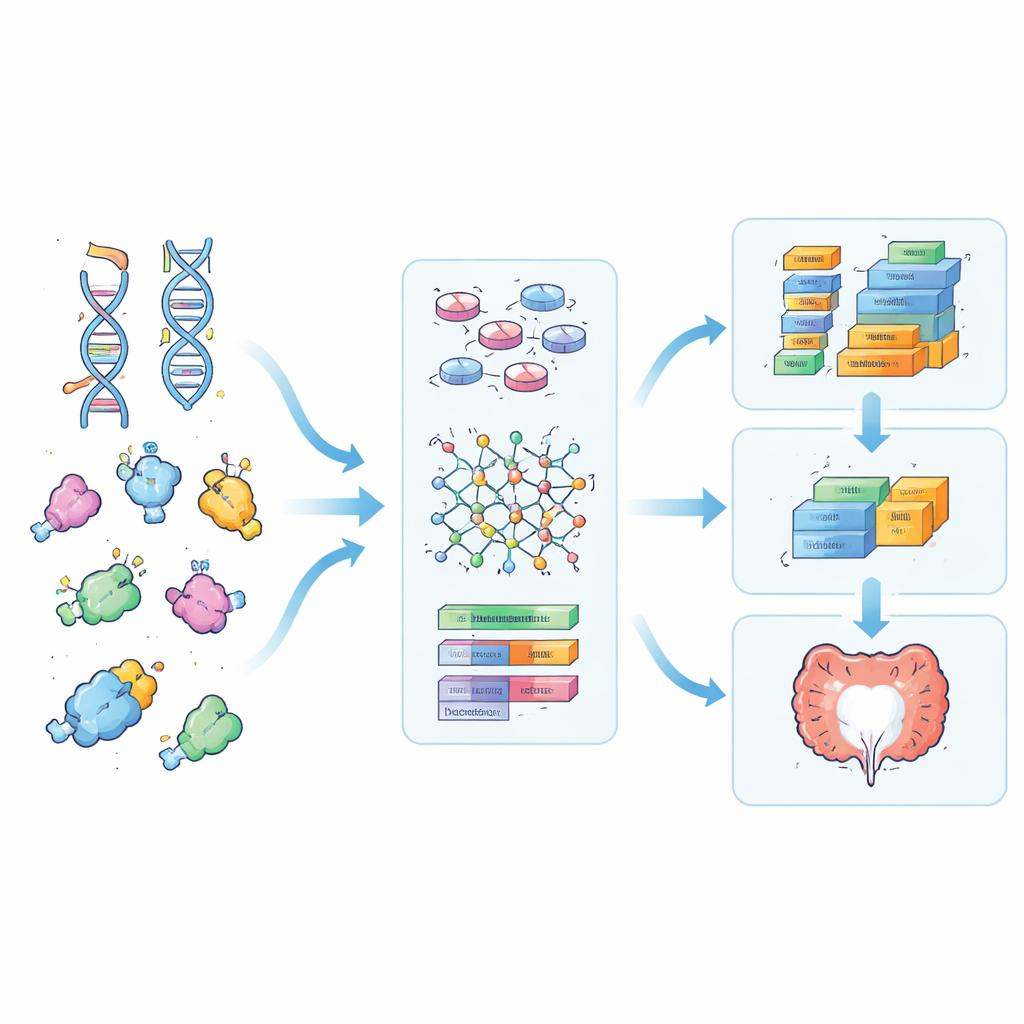
Misturando sinais genéticos com atividade gênica
Em seguida, a equipe adicionou a atividade gênica proveniente das medições de RNA, feitas no mesmo tecido intestinal usado nos testes do medicamento. Para cada proteína priorizada, emparelharam sua força de ligação prevista com a atividade de seu gene correspondente, e também multiplicaram ambos para criar um “estado de interação” que reflete tanto quão fortemente o medicamento é esperado se ligar quanto quanto do alvo está presente. Modelos de aprendizado de máquina treinados nessas características enriquecidas foram melhores em distinguir entre amostras que responderam bem à prednisolona e aquelas que não responderam, em comparação com modelos usando representações mais básicas dos mesmos dados de DNA. Algumas das características mais influentes apontaram para famílias de receptores relacionados ao olfato e um receptor envolvido no sinal calmante do cérebro, o GABA, sugerindo papéis subestimados dessas proteínas na inflamação intestinal e na resposta a esteróides.
Tornando ferramentas complexas utilizáveis por não especialistas
Além da biologia, os pesquisadores também enfrentaram uma barreira prática: a maioria dos laboratórios não tem a expertise computacional necessária para executar grandes modelos de base. Para resolver isso, eles empacotaram seu fluxo de trabalho de afinidade de ligação em um servidor de Protocolo de Contexto de Modelo (MCP) aberto que pode ser chamado diretamente por assistentes de IA como Claude ou ChatGPT. Com essa configuração, um cientista pode pedir, em linguagem simples, a predição de ligação entre um fármaco e um conjunto de proteínas, e o sistema lida discretamente com as etapas técnicas nos bastidores. Isso reduz a barreira para que outros investiguem questões semelhantes de resposta personalizada a medicamentos usando seus próprios dados.
O que isso significa para escolhas de tratamento futuras
Em termos práticos, este trabalho mostra como um único e poderoso modelo de IA treinado em enormes conjuntos de dados públicos pode ajudar estudos pequenos e do mundo real a alcançarem desempenho superior ao esperado. Ao transformar variações brutas de DNA e atividade gênica em características mais ricas e conscientes da biologia, os autores melhoraram a precisão de modelos que predizem quem tem maior probabilidade de se beneficiar da prednisolona. Os resultados ainda são iniciais e baseados em um número limitado de pacientes, de modo que os links específicos gene–fármaco que eles destacam precisarão de testes adicionais. Mas a estratégia geral — usar modelos de base como geradores inteligentes de características e torná-los acessíveis por meio de interfaces simples — aponta para um futuro em que médicos possam usar testes moleculares multicamadas, interpretados por IA, para escolher o medicamento e a dose certos para cada pessoa com DII.
Citação: Gardiner, LJ., Kelly, J., Evans, A. et al. Multi-omics feature engineering driven by biomedical foundation models improves drug response prediction for inflammatory bowel disease patients. Sci Rep 16, 13820 (2026). https://doi.org/10.1038/s41598-026-44366-y
Palavras-chave: doença inflamatória intestinal, predição de resposta a medicamentos, modelos de base biomédicos, multi-ômicas, medicina personalizada