Clear Sky Science · fr
L’ingénierie de caractéristiques multi-omiques guidée par des modèles fondamentaux biomédicaux améliore la prédiction de la réponse aux médicaments pour les patients atteints de maladies inflammatoires de l’intestin
Pourquoi cette recherche compte pour les patients
Les personnes atteintes de maladie inflammatoire de l’intestin (MII) font souvent l’expérience d’un parcours d’essais et d’erreurs frustrant lorsqu’elles commencent un nouveau traitement. Un même médicament peut calmer une inflammation intestinale sévère chez une personne et à peine soulager une autre. Cette étude examine comment de puissants nouveaux modèles d’intelligence artificielle (IA), initialement conçus pour comprendre la biologie à grande échelle, peuvent être réutilisés pour extraire beaucoup plus d’informations à partir de petits jeux de données patients. L’objectif est simple mais ambitieux : utiliser le profil génétique et moléculaire de chaque personne pour prédire à l’avance dans quelle mesure elle répondra à un médicament courant contre la MII.
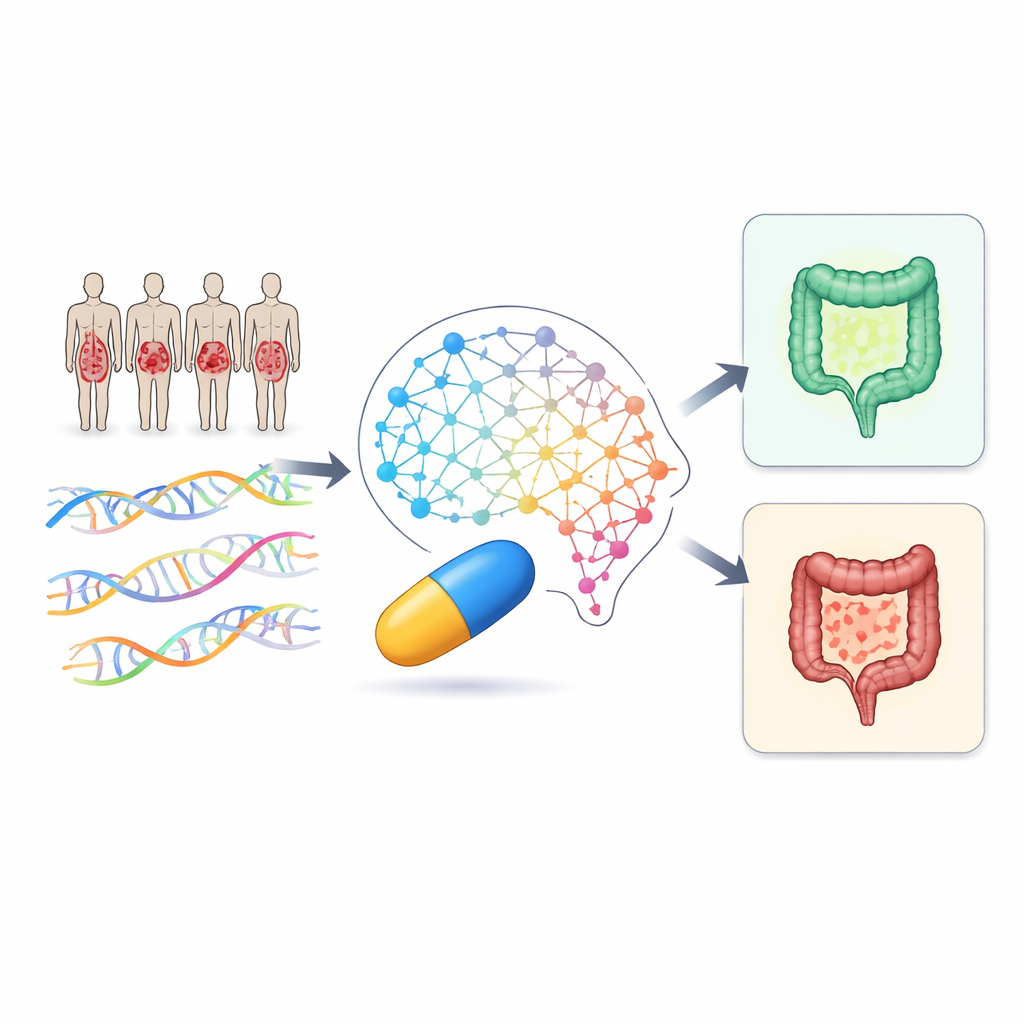
Utiliser une grande IA pour aider des études de petite taille
Les outils d’IA médicale traditionnels ont du mal lorsque le nombre de patients est faible, même si chaque patient est profondément caractérisé. Dans ce travail, les chercheurs renversent ce problème en puisant des connaissances dans un « modèle fondamental » massif appelé MAMMAL, entraîné à l’origine sur d’énormes collections de données de protéines et de médicaments. Plutôt que de réentraîner ce modèle, ils l’utilisent comme une calculatrice intelligente pour estimer la force d’interaction d’un médicament donné avec des milliers de protéines humaines. Ces scores d’affinité deviennent ensuite de nouvelles caractéristiques riches en information qui peuvent être intégrées à des modèles d’apprentissage automatique plus modestes, adaptés aux 51 patients atteints de MII de cette étude.
Regarder dans l’intestin au niveau de l’ADN et de l’ARN
L’équipe a travaillé sur des prélèvements de tissu prélevés dans les intestins enflammés de personnes atteintes de rectocolite hémorragique ou de maladie de Crohn. Pour chaque échantillon, ils ont mesuré deux couches biologiques principales : l’ADN, en se concentrant sur de petites variations génétiques appelées SNP, et l’ARN, qui reflète l’activité de chaque gène dans le tissu. Ils ont également exposé le tissu à la prédnisolone, un traitement stéroïdien de première ligne courant pour la MII, et mesuré dans quelle mesure un signal inflammatoire clé, le TNFα, diminuait en réponse. Ce changement de TNFα a servi de substitut pour évaluer la sensibilité du tissu du patient au médicament. Le défi consistait à relier des milliers de mesures génétiques et moléculaires à ce résultat de réponse au traitement.
Apprendre aux modèles ce à quoi prêter attention
La première étape a été de décider quelles protéines et quels gènes devaient retenir le plus l’attention des modèles. En utilisant le modèle fondamental, les chercheurs ont estimé à quel point la prédnisolone se lierait fortement à plus de 11 000 protéines humaines, puis se sont concentrés sur les 5 % supérieurs présentant la liaison prédite la plus forte. De manière encourageante, cette courte liste comprenait les cibles connues du médicament et des protéines impliquées dans la détection chimique et la signalisation, ce qui suggère que le classement de l’IA était biologiquement pertinent. Ils ont ensuite combiné cette liste avec les variations d’ADN de chaque patient, créant des séquences protéiques « mutées » personnalisées et recalculant comment chaque mutation pourrait légèrement renforcer ou affaiblir la liaison de la prédnisolone. Ces variations prédictives d’affinité sont devenues des caractéristiques compactes et spécifiques au patient, capturant comment des variants dans des protéines clés pourraient modifier l’action du médicament.
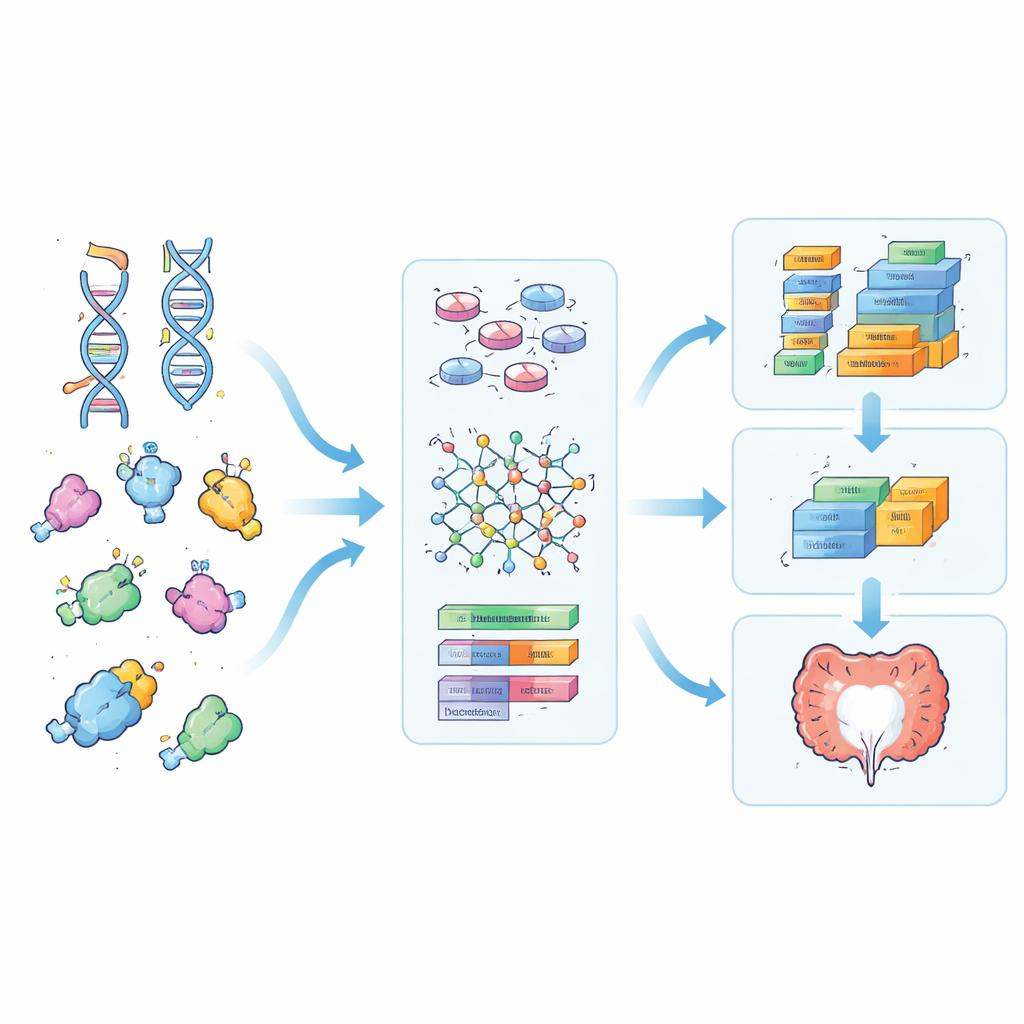
Mélanger signaux génétiques et activité génique
Ensuite, l’équipe a superposé l’activité génique issue des mesures d’ARN, prélevées dans le même tissu intestinal utilisé pour les tests médicamenteux. Pour chaque protéine priorisée, ils ont associé la force de liaison prédite à l’activité de son gène correspondant, et ont aussi multiplié les deux pour créer un « état d’interaction » reflétant à la fois la force d’affinité attendue du médicament et la quantité de cible présente. Les modèles d’apprentissage automatique entraînés sur ces caractéristiques enrichies étaient meilleurs pour distinguer les échantillons répondant bien à la prédnisolone de ceux qui ne répondaient pas, comparativement à des modèles utilisant des représentations plus basiques des mêmes données d’ADN. Certaines des caractéristiques les plus influentes désignaient des familles de récepteurs liés à l’odorat et un récepteur impliqué dans le signal calmant GABA, suggérant des rôles sous-estimés de ces protéines dans l’inflammation intestinale et la réponse aux stéroïdes.
Rendre des outils complexes utilisables par des non-experts
Au-delà de la biologie, les chercheurs ont aussi abordé une barrière pratique : la plupart des laboratoires n’ont pas l’expertise informatique nécessaire pour exécuter de grands modèles fondamentaux. Pour y remédier, ils ont encapsulé leur flux de travail d’affinité de liaison dans un serveur ouvert de Model Context Protocol (MCP) qui peut être sollicité directement par des assistants IA tels que Claude ou ChatGPT. Avec ce dispositif, un scientifique peut demander, en langage simple, la prédiction de liaison entre un médicament et un ensemble de protéines, et le système gère discrètement les étapes techniques en arrière-plan. Cela abaisse le seuil pour que d’autres explorent des questions similaires de réponse médicamenteuse personnalisée avec leurs propres données.
Ce que cela signifie pour les choix thérapeutiques futurs
Concrètement, ce travail montre comment un modèle IA unique et puissant, entraîné sur d’énormes jeux de données publics, peut permettre à de petites études cliniques réelles d’avoir un impact supérieur à leur taille. En transformant des variations d’ADN brutes et l’activité génique en caractéristiques plus riches et informées par la biologie, les auteurs ont amélioré la précision des modèles qui prédisent qui est susceptible de bénéficier de la prédnisolone. Les résultats sont encore préliminaires et basés sur un nombre limité de patients, si bien que les liens gène–médicament spécifiques mis en avant nécessiteront des validations supplémentaires. Mais la stratégie générale — utiliser des modèles fondamentaux comme générateurs intelligents de caractéristiques et les rendre accessibles via des interfaces simples — ouvre la voie à un avenir où les médecins pourraient utiliser des tests moléculaires multi-couches, interprétés par l’IA, pour choisir le bon médicament et la bonne posologie pour chaque personne atteinte de MII.
Citation: Gardiner, LJ., Kelly, J., Evans, A. et al. Multi-omics feature engineering driven by biomedical foundation models improves drug response prediction for inflammatory bowel disease patients. Sci Rep 16, 13820 (2026). https://doi.org/10.1038/s41598-026-44366-y
Mots-clés: maladie inflammatoire de l’intestin, prédiction de la réponse aux médicaments, modèles fondamentaux biomédicaux, multi-omiques, médecine personnalisée