Clear Sky Science · pt
Mobilidades de portadores e interações elétron-fônon além do DFT
Por que átomos que se agitam importam para a eletrônica
Todo sólido está cheio de átomos inquietos que vibram, mesmo à temperatura ambiente. Esses pequenos tremores colidem constantemente com os elétrons que transportam corrente elétrica, estabelecendo um limite fundamental para a velocidade com que dispositivos podem operar e para a eficiência com que usam energia. Este artigo apresenta uma nova maneira de calcular, a partir de primeiros princípios, como essas vibrações atômicas desaceleram elétrons e lacunas em semicondutores importantes como silício e arseneto de gálio, utilizando alguns dos métodos de estrutura eletrônica mais precisos disponíveis hoje.

Como elétrons e vibrações se comunicam
No interior de um cristal, elétrons movem-se por um panorama periódico criado pelos átomos, enquanto os próprios átomos vibram em padrões coletivos chamados fônons. Quando os dois interagem, elétrons podem espalhar-se, mudando sua energia e direção. Esse acoplamento elétron–fônon governa propriedades-chave, como a facilidade de fluxo de carga (mobilidade), a intensidade da absorção de luz e até se um material se torna supercondutor. Cálculos tradicionais desse acoplamento baseiam-se na teoria do funcional da densidade (DFT), um método de grande sucesso porém aproximado. Embora as ferramentas baseadas em DFT estejam bastante maduras, elas ainda têm dificuldade em reproduzir experimentos para alguns materiais, especialmente quando descrições mais precisas das excitações eletrônicas são necessárias.
Indo além das receitas eletrônicas padrão
Para melhorar o DFT padrão, pesquisadores usam métodos avançados de estrutura eletrônica, incluindo funcionais híbridos, funcionais compatíveis com Koopmans e técnicas de muitos corpos GW. Essas abordagens corrigem deficiências antigas do DFT, como lacunas de banda subestimadas e interações eletrônicas excessivamente amortecidas, e em geral fornecem energias de quasipartículas melhores. Contudo, combiná-las diretamente com as técnicas existentes de elétron–fônon é difícil. Seus potenciais efetivos são mais complexos, podem depender de orbitais individuais e frequentemente são não locais e dependentes de frequência, tornando os esquemas perturbativos padrão difíceis de implementar e muito custosos em tempo de computador e memória.
Uma nova rota usando deslocamentos de energia em vez de potenciais
Os autores introduzem um framework de diferenças finitas que contorna essas dificuldades ao focar nos níveis de energia e nas funções de onda em vez do potencial microscópico completo. Eles calculam como as energias eletrônicas mudam quando os átomos em uma supercélula são ligeiramente deslocados e então reconstruem os elementos matriciais elétron–fônon usando um esquema de “projetabilidade” baseado em sobreposições entre estados perturbados e não perturbados. O uso inteligente de simetria reduz drasticamente o número de deslocamentos atômicos independentes que precisam ser calculados. O fluxo de trabalho foi concebido para se integrar a códigos amplamente usados, como Quantum ESPRESSO, KOOPMANS e YAMBO, e então repassar os acoplamentos resultantes para o código EPW, que usa funções de Wannier para interpolá-los em grades de momento extremamente finas necessárias para cálculos de transporte precisos.
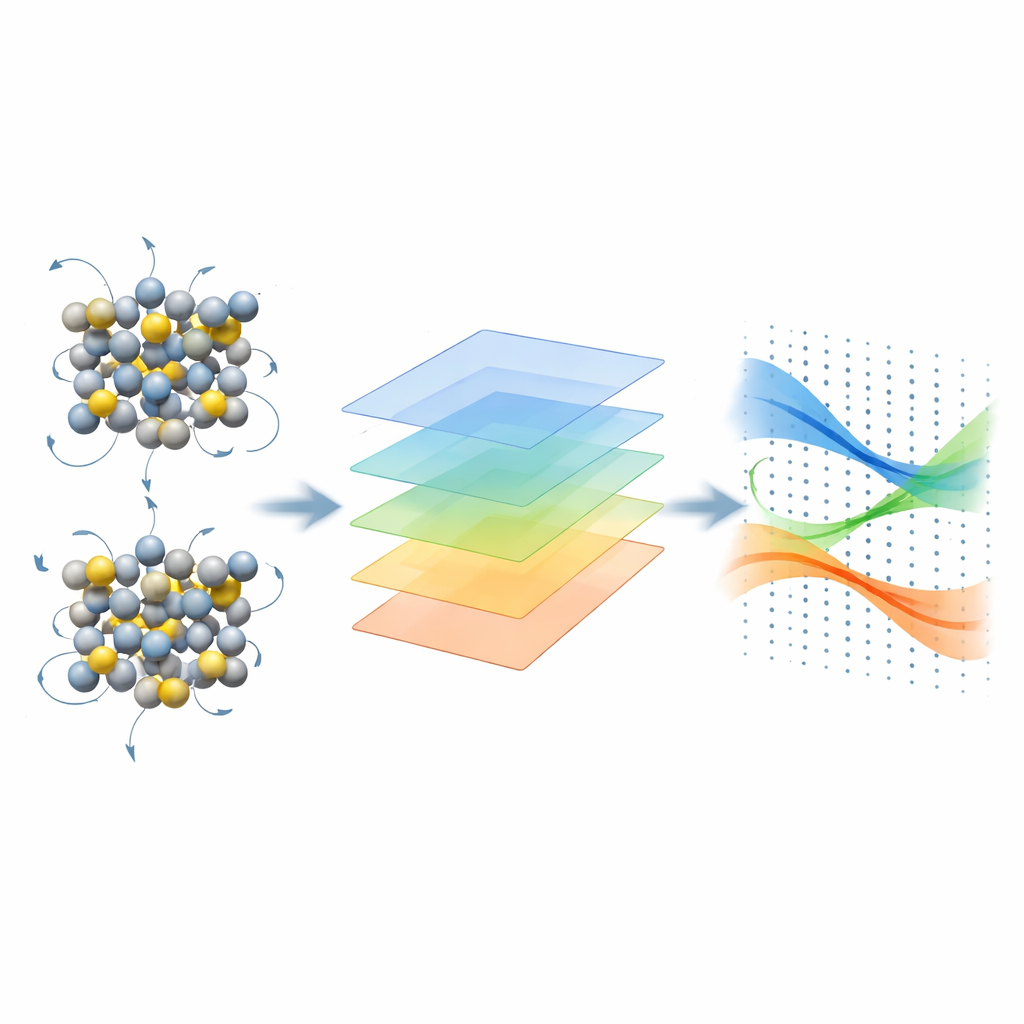
O que muda quando elétrons são tratados de forma mais realista
Com essa máquina em funcionamento, a equipe examina silício e arseneto de gálio, dois semicondutores amplamente utilizados. Eles comparam o DFT padrão com métodos avançados para calcular o acoplamento elétron–fônon, a curvatura das bandas de energia (que determina a massa efetiva) e a mobilidade de deriva resultante, obtida a partir de uma solução detalhada da equação de transporte de Boltzmann. No silício, os métodos mais refinados aumentam ligeiramente a força do acoplamento elétron–fônon e ajustam a curvatura das bandas, levando a reduções modestas nas mobilidades de elétrons e lacunas — da ordem de 10% — aproximando ainda mais a teoria das medições. No arseneto de gálio, onde se sabe que o DFT subestima a massa efetiva do elétron e superestima a mobilidade, os funcionais híbridos e de Koopmans corrigem a curvatura das bandas e aumentam moderadamente o acoplamento, reduzindo a mobilidade eletrônica prevista de valores irrealisticamente altos para números que se alinham bem com experimentos em uma ampla faixa de temperaturas.
Uma imagem mais clara para projetar materiais melhores
Para não especialistas, a mensagem principal é que prever com precisão o quão bem um material conduz eletricidade exige acertar tanto os elétrons quanto as vibrações atômicas — e, crucialmente, como eles se influenciam mutuamente. Este trabalho entrega um arcabouço geral e prático para fazer exatamente isso, usando métodos de estrutura eletrônica de ponta sem implementações específicas para cada nova abordagem. Empacotado em um novo código chamado ElePhAny e interconectado com ferramentas estabelecidas, o método abre a porta para cálculos de alta precisão rotineiros de mobilidade e outras propriedades dirigidas por elétron–fônon em uma ampla gama de materiais. Isso, por sua vez, pode orientar a descoberta e otimização de materiais eletrônicos e optoeletrônicos antes mesmo de serem sintetizados em laboratório.
Citação: Poliukhin, A., Colonna, N., Libbi, F. et al. Carrier mobilities and electron-phonon interactions beyond DFT. npj Comput Mater 12, 151 (2026). https://doi.org/10.1038/s41524-026-02011-2
Palavras-chave: acoplamento elétron-fônon, mobilidade de portadores, semicondutores, além do DFT, transporte a partir de primeiros princípios