Clear Sky Science · es
Movilidades de portadores e interacciones electrón-fonón más allá del DFT
Por qué importan los átomos que vibran para la electrónica
Cualquier sólido está lleno de átomos inquietos que vibran, incluso a temperatura ambiente. Estos sutiles sacudimientos chocan continuamente con los electrones que transportan la corriente eléctrica, estableciendo un límite fundamental a la rapidez con la que pueden operar los dispositivos y a la eficiencia en el uso de energía. Este artículo presenta una nueva forma de calcular, desde primeros principios, cómo estas vibraciones atómicas frenan a electrones y huecos en semiconductores relevantes como el silicio y el arseniuro de galio, utilizando algunos de los métodos de estructura electrónica más precisos disponibles hoy.

Cómo se comunican electrones y vibraciones
Dentro de un cristal, los electrones se desplazan por un paisaje periódico creado por los átomos, mientras que los propios átomos vibran en patrones colectivos llamados fonones. Cuando ambos interactúan, los electrones pueden dispersarse, cambiando su energía y dirección. Este acoplamiento electrón–fonón gobierna propiedades clave como la facilidad con la que fluye la carga (movilidad), la intensidad con que se absorbe la luz e incluso si un material se vuelve superconductor. Los cálculos tradicionales de este acoplamiento se basan en la teoría del funcional de la densidad (DFT), un método muy exitoso pero aproximado. Aunque las herramientas basadas en DFT están muy desarrolladas, aun así les cuesta reproducir experimentos en algunos materiales, especialmente cuando se requieren descripciones más precisas de las excitaciones electrónicas.
Ir más allá de las recetas electrónicas estándar
Para mejorar sobre el DFT estándar, los investigadores usan métodos de estructura electrónica más avanzados, incluidos funcionales híbridos, funcionales compatibles con Koopmans y técnicas many-body GW. Estos enfoques corrigen deficiencias de larga data del DFT, como la subestimación de las brechas de banda y el sobredetenimiento de las interacciones electrónicas, y en general proporcionan mejores energías de cuasipartículas. Sin embargo, combinarlos directamente con las técnicas existentes de electrón–fonón es difícil. Sus potenciales efectivos son más complejos, pueden depender de orbitales individuales y con frecuencia son no locales y dependientes de la frecuencia, lo que hace que los esquemas perturbativos estándar sean difíciles de implementar y muy costosos en tiempo de cálculo y memoria.
Una nueva vía que usa desplazamientos de energía en lugar de potenciales
Los autores introducen un marco de diferencia finita que evita estas dificultades al centrarse en los niveles de energía y las funciones de onda en lugar del potencial microscópico completo. Calculan cómo cambian las energías electrónicas cuando se desplazan ligeramente los átomos en una supercelda y luego reconstruyen los elementos matriciales electrón–fonón usando un esquema de “proyectabilidad” basado en solapamientos entre estados perturbados y no perturbados. El uso inteligente de la simetría reduce drásticamente cuántos desplazamientos atómicos independientes hay que calcular. El flujo de trabajo está diseñado para integrarse con códigos ampliamente usados como Quantum ESPRESSO, KOOPMANS y YAMBO, y después pasa los acoplamientos resultantes al código EPW, que emplea funciones de Wannier para interpolarlos en rejillas de momento extremadamente finas necesarias para cálculos de transporte precisos.
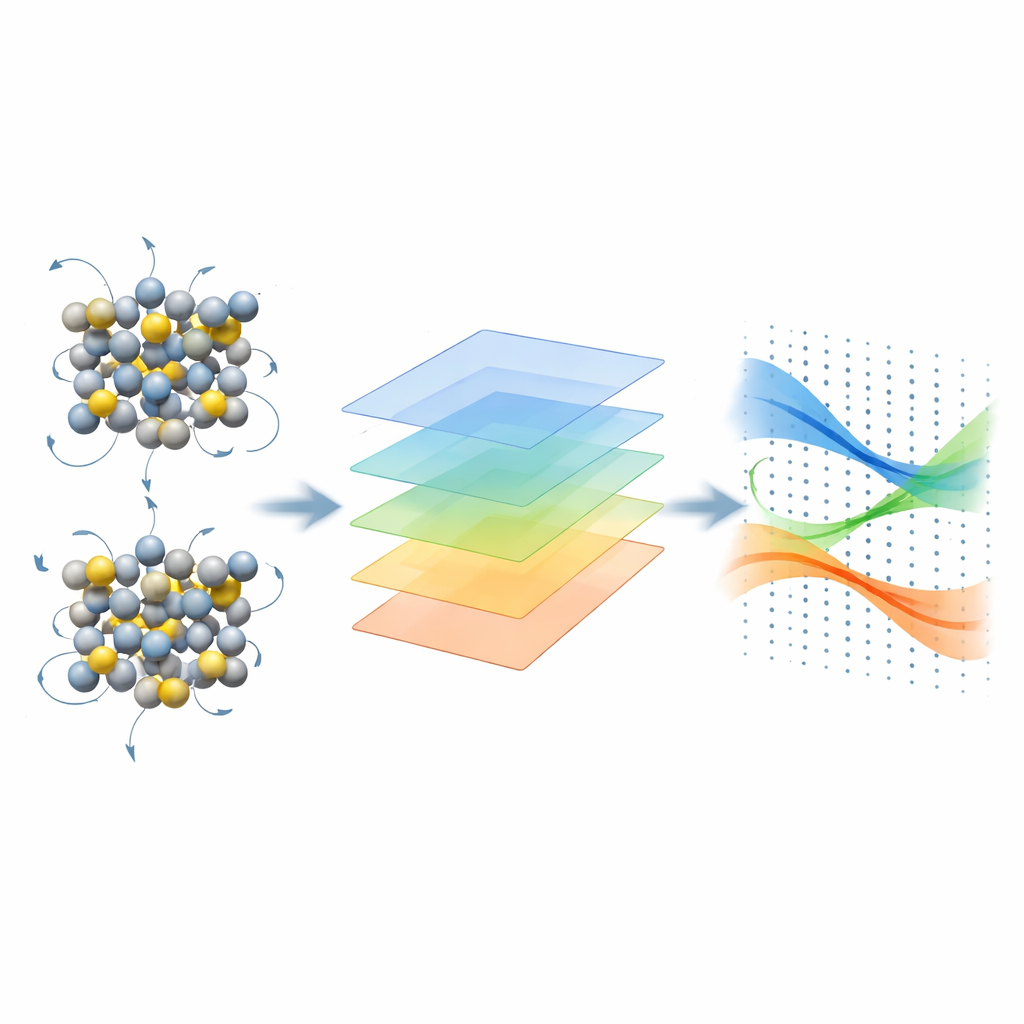
Qué cambia cuando los electrones se tratan de forma más realista
Con esta maquinaria en marcha, el equipo examina el silicio y el arseniuro de galio, dos semiconductores fundamentales. Comparan el DFT estándar con métodos avanzados para calcular el acoplamiento electrón–fonón, la curvatura de las bandas de energía (que determina la masa efectiva) y la movilidad de deriva resultante obtenida a partir de una solución detallada de la ecuación de transporte de Boltzmann. En el silicio, los métodos más refinados aumentan ligeramente la intensidad del acoplamiento electrón–fonón y ajustan la curvatura de las bandas, lo que conduce a reducciones modestas tanto de la movilidad de electrones como de huecos — del orden del 10% — que acercan la teoría aún más a las medidas experimentales. En el arseniuro de galio, donde se sabe que el DFT subestima la masa efectiva del electrón y sobrestima la movilidad, los funcionales híbridos y de Koopmans corrigen la curvatura de bandas y aumentan moderadamente el acoplamiento, reduciendo la movilidad electrónica predicha desde valores irrealmente altos hasta cifras que se alinean bien con los experimentos en un amplio rango de temperaturas.
Una visión más clara para diseñar mejores materiales
Para no especialistas, el mensaje clave es que predecir con precisión cuán bien conduce electricidad un material requiere acertar tanto en los electrones como en las vibraciones atómicas y, de forma crucial, en cómo se influyen mutuamente. Este trabajo ofrece un marco general y práctico para hacer exactamente eso, usando métodos de estructura electrónica de vanguardia sin implementaciones a medida para cada nuevo enfoque. Empaquetado en un nuevo código llamado ElePhAny e integrado con herramientas consolidadas, el método abre la puerta a cálculos de movilidad y otras propiedades gobernadas por el electrón–fonón de alta precisión y de uso rutinario en una amplia gama de materiales. Esto, a su vez, puede orientar el descubrimiento y la optimización de materiales electrónicos y optoelectrónicos antes de que se fabriquen en el laboratorio.
Cita: Poliukhin, A., Colonna, N., Libbi, F. et al. Carrier mobilities and electron-phonon interactions beyond DFT. npj Comput Mater 12, 151 (2026). https://doi.org/10.1038/s41524-026-02011-2
Palabras clave: acoplamiento electrón-fonón, movilidad de portadores, semiconductores, más allá del DFT, transporte desde primeros principios