Clear Sky Science · pt
Solução equivariante por difusão para determinação de estruturas cristalinas inorgânicas a partir de dados de difração de raios X em pó
Ensinando Computadores a Ler Impressões Digitais Cristalinas
Muitas das tecnologias atuais, de baterias a catalisadores e ímãs, dependem do arranjo preciso dos átomos dentro de cristais inorgânicos. Essa arquitetura invisível normalmente é decodificada por raios X, que deixam um padrão distintivo — uma “impressão digital”. Mas transformar esses padrões em um mapa atômico preciso tem exigido anos de treinamento e tentativas demoradas. Este estudo apresenta um sistema de IA, chamado XRDSol, capaz de ler essas impressões e propor estruturas cristalinas completas em menos de um segundo, abrindo caminho para descoberta de materiais mais rápida e bancos de dados de materiais mais confiáveis.
Por Que Padrões em Pó São Tão Difíceis de Decifrar
Quando raios X atravessam um cristal único perfeitamente formado, eles criam um padrão tridimensional rico que pode ser usado para localizar cada átomo. Amostras do mundo real, contudo, frequentemente consistem de pós compostos por muitos grãos minúsculos. Seu padrão de difração colapsa para uma série unidimensional de picos, na qual grande parte da informação espacial original se perde. Especialistas humanos tipicamente precisam combinar esse padrão comprimido com seu conhecimento de química e cristalografia, estimando parâmetros de rede, simetria e posições atômicas, e então refinando suas hipóteses repetidamente. Para materiais complexos ou pouco conhecidos, as soluções podem ser incompletas, controversas ou mesmo incorretas, e grandes bancos de dados estruturais contêm milhares de entradas com coordenadas atômicas ausentes ou implausíveis.
Uma IA que Reconstrói Arranjos Atômicos
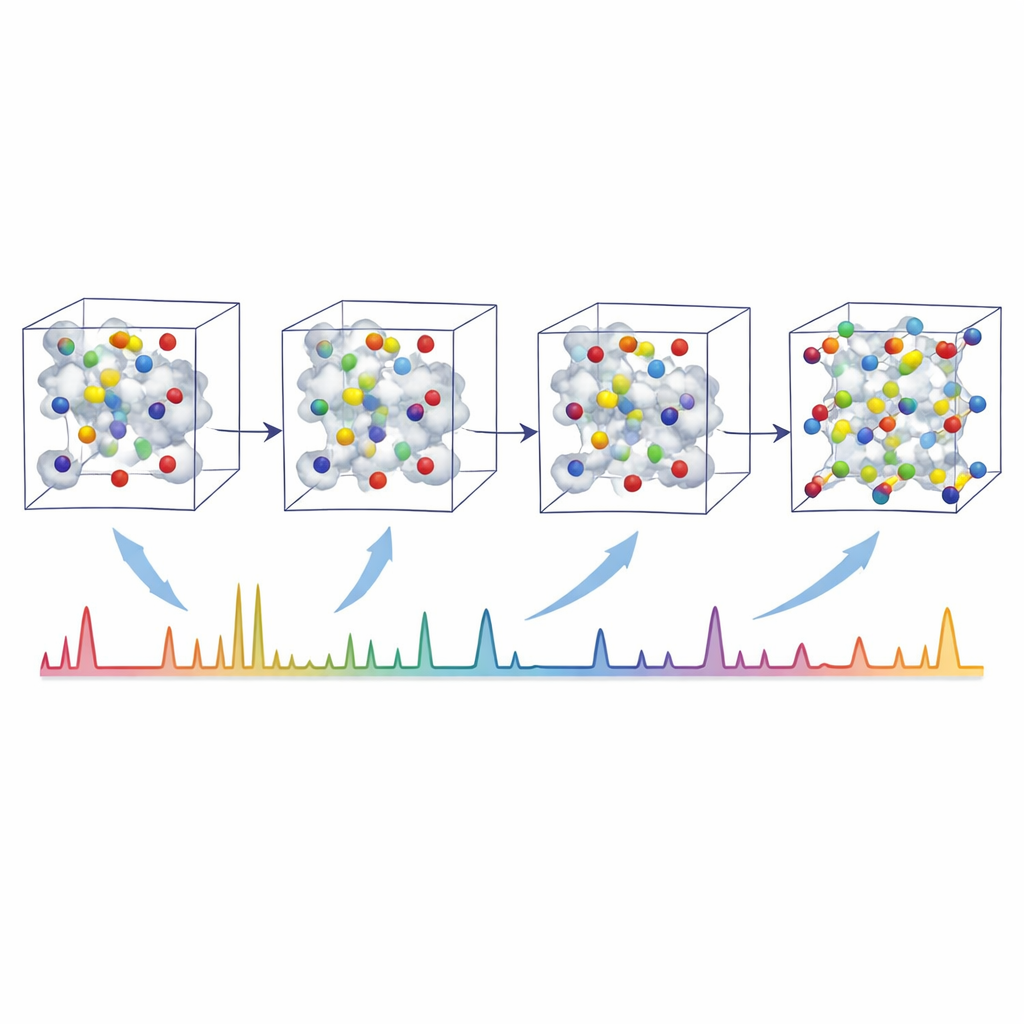
Os autores enfrentam esse desafio com o XRDSol, um modelo de inteligência artificial baseado em um processo de difusão equivariante atuando sobre uma representação em grafo do cristal. Em vez de começar a partir de um bom palpite, o XRDSol inicia com átomos colocados em posições aleatórias dentro de uma cela unitária conhecida (com fórmula química e parâmetros de rede conhecidos). Durante o treinamento, o modelo aprende a reverter um gradual processo de “ruído” no qual estruturas estáveis e termodinamicamente favorecidas são repetidamente distorcidas. Guiado por uma representação compacta do padrão de difração de raios X em pó alvo, o modelo “desruidosa” iterativamente o arranjo aleatório, conduzindo os átomos passo a passo para posições que são tanto quimicamente plausíveis quanto consistentes com o padrão observado. Como a rede neural subjacente respeita as simetrias rotacionais e translacionais dos cristais, ela naturalmente favorece arranjos fisicamente razoáveis.
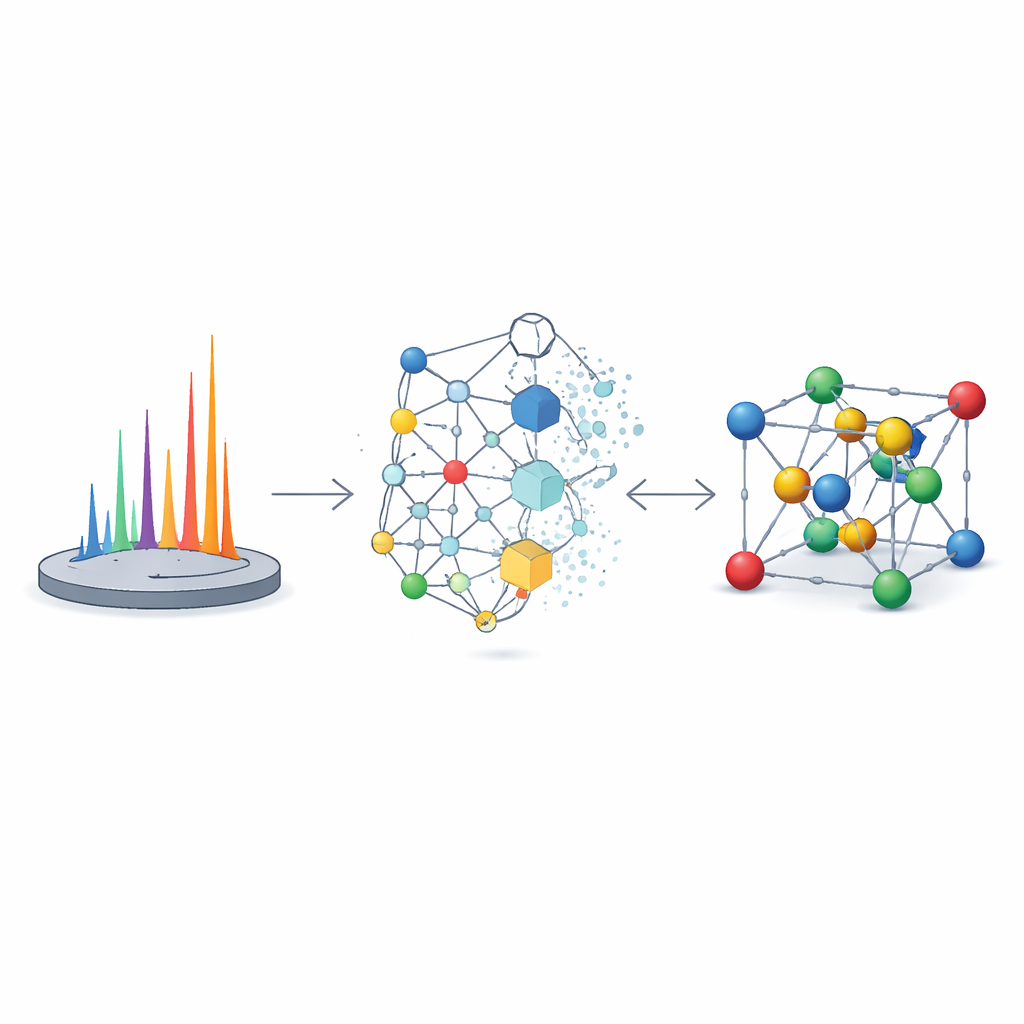
Soluções Rápidas e Precis
Para testar o XRDSol, a equipe usou um conjunto de dados com mais de 9.000 estruturas inorgânicas estáveis com padrões de pó simulados. Em uma única placa gráfica, o modelo leva cerca de 0,6 segundos para gerar uma solução para uma estrutura — aproximadamente de dez mil a cem mil vezes mais rápido que métodos anteriores que dependem de cálculos quânticos pesados e buscas evolucionárias. Em mais de 80% dos casos, o XRDSol recuperou posições atômicas que correspondem de perto às estruturas conhecidas, e em mais de 90% a padrão de difração reconstruído foi altamente semelhante ao alvo. O método funciona especialmente bem para cristais de alta simetria, embora o desempenho caia para casos de baixa simetria e mais complexos. Ainda assim, exemplos que vão de sais simples a óxidos, sulfetos e compostos intermetálicos complexos mostram que a abordagem é amplamente aplicável a diferentes químicas.
Corrigindo Registros Antigos e Completando Estruturas Ausentes
Além de reproduzir respostas conhecidas, o XRDSol pode melhorar resultados duvidosos. Os autores revisitaram milhares de entradas de banco de dados com energias calculadas anormalmente altas — um indicativo de que algo está errado na estrutura publicada. Usando apenas o padrão de pó, a cela unitária e a composição como entrada, o XRDSol propôs arranjos atômicos alternativos. Para pelo menos 39 compostos, as novas estruturas corresponderam melhor aos dados de difração e apresentaram energias muito menores, concordando com re-determinações experimentais posteriores em vários casos bem estudados. O sistema também completou coordenadas faltantes para 912 entradas cujos padrões de difração eram conhecidos, mas cujas posições atômicas estavam ausentes, incluindo exemplos desafiadores contendo elementos leves como hidrogênio e lítio, minerais naturais com impurezas e materiais exibindo desordem química. Essas estruturas geradas pela IA foram verificadas com cálculos quânticos e inspeção manual e mostraram-se energeticamente plausíveis e quimicamente razoáveis.
Rumo à Descoberta Automatizada de Materiais
O XRDSol demonstra que uma rede neural sensível à simetria baseada em difusão pode aprender grande parte do conhecimento de especialista necessário para resolver estruturas cristalinas inorgânicas diretamente a partir de dados de difração de raios X em pó. Embora o método ainda tenha dificuldades com celas unitárias muito grandes, fases de baixa simetria e sítios completamente desordenados, ele já fornece modelos iniciais rápidos e de alta qualidade para refinamento adicional. Em termos práticos, isso significa análises de rotina mais rápidas para não especialistas, uma ferramenta poderosa para limpar e completar grandes bancos de dados estruturais e um componente-chave para laboratórios em circuito fechado onde computadores projetam, sintetizam, caracterizam e otimizam novos materiais com intervenção humana mínima.
Citação: Yu, D., Zhu, Z., Leng, F. et al. Equivariant diffusion solution for inorganic crystal structure determination from powder X-ray diffraction data. Nat Commun 17, 3274 (2026). https://doi.org/10.1038/s41467-026-70035-9
Palavras-chave: difração de raios X em pó, determinação de estrutura cristalina, modelo de difusão equivariante, informática de materiais, redes neurais gráficas