Clear Sky Science · en
Equivariant diffusion solution for inorganic crystal structure determination from powder X-ray diffraction data
Teaching Computers to Read Crystal Fingerprints
Many of today’s technologies, from batteries to catalysts and magnets, rely on the precise arrangement of atoms inside inorganic crystals. That invisible architecture is usually decoded using X-rays, which leave behind a distinctive “fingerprint” pattern. But turning those patterns into an accurate atomic map has long required years of training and painstaking trial-and-error. This study introduces an AI system, called XRDSol, that can read these fingerprints and propose full crystal structures in less than a second, opening the door to faster materials discovery and more reliable materials databases.
Why Powder Patterns Are So Hard to Crack
When X-rays pass through a perfectly shaped single crystal, they create a rich three-dimensional pattern that can be used to pinpoint each atom. Real-world samples, however, are often powders made of many tiny grains. Their X-ray diffraction pattern collapses into a one-dimensional series of peaks, where much of the original spatial information is lost. Human experts typically have to combine this compressed pattern with their knowledge of chemistry and crystallography, guessing lattice parameters, symmetry, and atomic positions, then refining their guesses repeatedly. For complex or poorly known materials, solutions may be incomplete, controversial, or even wrong, and large structural databases contain thousands of entries with missing or implausible atomic coordinates.
An AI That Reconstructs Atomic Arrangements
The authors tackle this challenge with XRDSol, an artificial intelligence model based on an equivariant diffusion process acting on a graph representation of the crystal. Instead of starting from a good guess, XRDSol begins with atoms placed at random positions inside a known unit cell (with known chemical formula and lattice parameters). During training, the model learns to reverse a gradual “noising” process in which well-established, thermodynamically stable structures are repeatedly distorted. Guided by a compressed representation of the target powder X-ray diffraction pattern, the model iteratively “denoises” the random arrangement, nudging atoms step by step into positions that are both chemically sensible and consistent with the observed pattern. Because the underlying neural network respects the rotational and translational symmetries of crystals, it naturally favors physically reasonable arrangements.
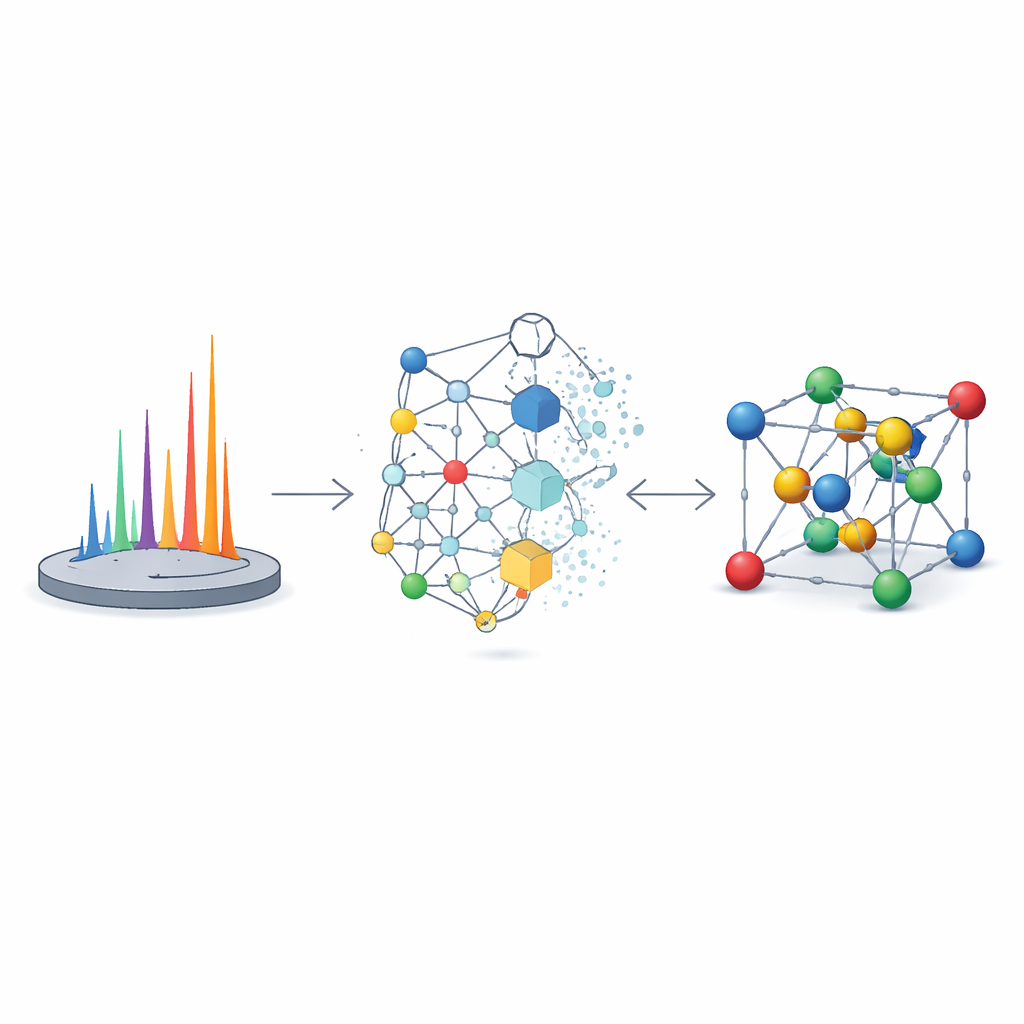
Fast, Accurate Solutions Across Many Materials
To test XRDSol, the team used a dataset of over 9,000 stable inorganic structures with simulated powder patterns. On a single graphics processor, the model takes about 0.6 seconds to generate a solution for one structure—roughly ten thousand to one hundred thousand times faster than earlier methods that rely on heavy quantum-mechanical calculations and evolutionary searches. In more than 80 percent of cases, XRDSol recovered atomic positions that closely match the known structures, and in over 90 percent, the reconstructed diffraction pattern was highly similar to the target. The method works especially well for highly symmetric crystals, though performance drops for low-symmetry, more complicated cases. Still, examples ranging from simple salts to complex oxides, sulfides, and intermetallic compounds show that the approach is broadly applicable across different chemistries.
Correcting Old Records and Completing Missing Structures
Beyond reproducing known answers, XRDSol can improve questionable ones. The authors revisited thousands of database entries with unusually high calculated energies—an indicator that something is wrong with the published structure. Using only the powder pattern, lattice, and composition as input, XRDSol proposed alternative atomic arrangements. For at least 39 compounds, the new structures better matched the diffraction data and had far lower energies, agreeing with later experimental re-determinations in several well-studied cases. The system also filled in missing coordinates for 912 entries whose diffraction patterns were known but whose atomic positions were absent, including challenging examples containing light elements such as hydrogen and lithium, natural minerals with impurities, and materials exhibiting chemical disorder. These AI-generated structures were checked with quantum calculations and manual inspection and were found to be energetically plausible and chemically reasonable.
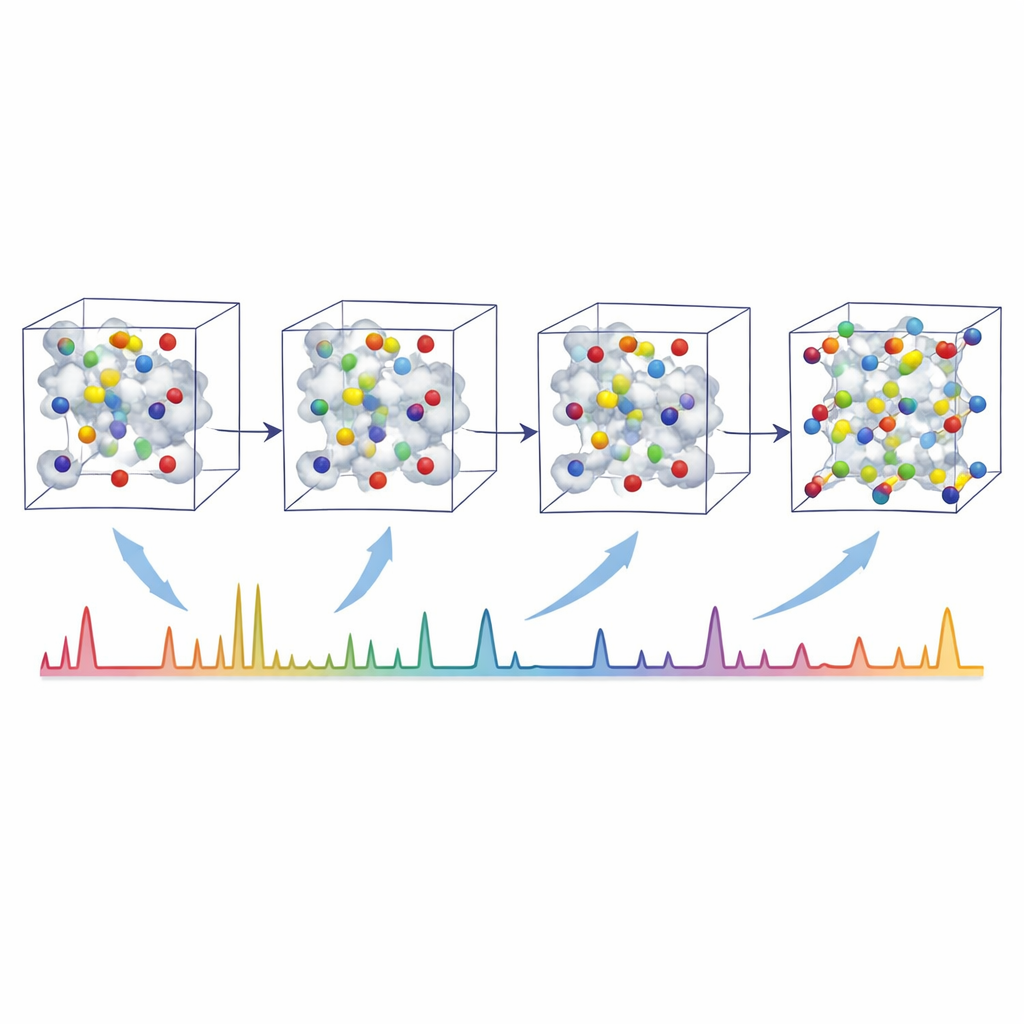
Toward Automated Materials Discovery
XRDSol demonstrates that a diffusion-based, symmetry-aware neural network can learn much of the expert knowledge needed to solve inorganic crystal structures directly from powder X-ray data. While the method still struggles with very large unit cells, low-symmetry phases, and fully disordered sites, it already provides rapid, high-quality starting models for further refinement. In practical terms, this means faster routine analysis for non-specialists, a powerful tool for cleaning and completing large structural databases, and a key component for closed-loop laboratories where computers design, synthesize, characterize, and optimize new materials with minimal human intervention.
Citation: Yu, D., Zhu, Z., Leng, F. et al. Equivariant diffusion solution for inorganic crystal structure determination from powder X-ray diffraction data. Nat Commun 17, 3274 (2026). https://doi.org/10.1038/s41467-026-70035-9
Keywords: powder X-ray diffraction, crystal structure determination, equivariant diffusion model, materials informatics, graph neural networks