Clear Sky Science · pt
Intermediários da formação de heteroestruturas de dicaniuretos de metais de transição revelados por simulações com aprendizado de máquina
Por que empilhar materiais ultrafinos é tão difícil
Eletrônica construída a partir de folhas com apenas alguns átomos de espessura promete dispositivos mais rápidos e eficientes. Uma família popular desses semicondutores ultrafinos é composta por metais ligados ao enxofre ou selênio, e quando diferentes folhas são empilhadas, elas podem se comportar como materiais inteiramente novos. Mas crescer pilhas grandes e perfeitas no laboratório tem se mostrado difícil: as camadas tendem a se misturar em ligas em vez de permanecem claramente separadas. Este estudo usa simulações computacionais avançadas alimentadas por aprendizado de máquina para observar as etapas ocultas de como essas pilhas crescem, revelando uma estrutura intermediária inesperada que ajuda a explicar tanto os problemas quanto as novas oportunidades para dispositivos.
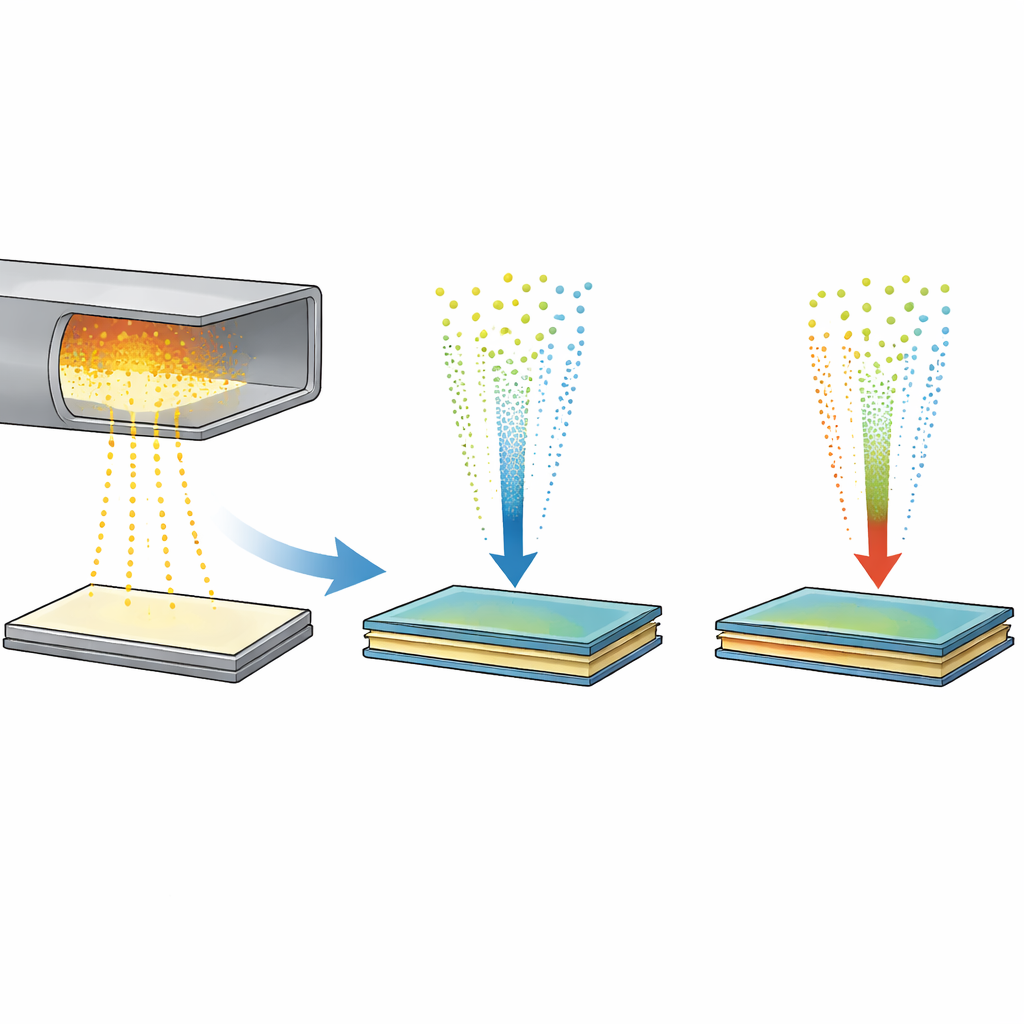
Construindo pilhas atômicas para os chips do futuro
Engenheiros têm especial interesse em empilhar materiais como dissulfeto de molibdênio (MoS2) e dissulfeto de tungstênio (WS2). Essas chamadas heteroestruturas van der Waals conduzem eletricidade de forma eficaz, interagem fortemente com a luz e podem ser montadas como peças de Lego em escala atômica. O empilhamento mecânico pode produzir interfaces nítidas e bem definidas, mas apenas em flocos minúsculos e a alto custo. Métodos escaláveis como deposição por vapor químico podem crescer camadas únicas por uma pastilha inteira, entretanto, quando pesquisadores tentam empilhar camadas diferentes, os metais tendem a trocar de lugar e formar ligas mistas, comprometendo o comportamento eletrônico limpo que os dispositivos exigem.
Usando simulações inteligentes como uma câmera atômica
Observar átomos se moverem durante o crescimento em um forno real é quase impossível, então os autores construíram um modelo digital altamente preciso. Eles treinaram um potencial de aprendizado de máquina — um modelo de inteligência artificial ajustado com milhares de cálculos quântico‑mecânicos — para imitar como átomos de molibdênio, tungstênio e enxofre interagem. Integrado a simulações de dinâmica molecular, esse modelo permitiu rastrear milhões de movimentos atômicos ao longo de nanossegundos mantendo precisão próxima à quântica. Verificaram que o modelo reproduzia fielmente estruturas, energias e vibrações conhecidas, garantindo que suas previsões sobre vias de crescimento são confiáveis.
Uma camada metálica enterrada que muda tudo
As simulações inicialmente investigaram o que acontece quando átomos metálicos nus pousam sobre uma folha existente de MoS2 ou WS2, espelhando um processo vapor em duas etapas usado em experimentos. Em vez de permanecerem por cima como um filme ordenado, átomos isolados de molibdênio rapidamente se infiltraram na camada de enxofre abaixo da superfície, formando uma camada metálica enterrada sanduichada entre folhas de enxofre — rotulada SMoMoS quando apenas molibdênio está envolvido, e SMMS quando molibdênio e tungstênio se misturam. Essa camada afundada é surpreendentemente estável e incentiva átomos metálicos em posições diferentes a trocar, o que conduz naturalmente à formação de ligas em vez de uma pilha limpa de MoS2/WS2. Em temperaturas mais baixas a troca desacelera, mas a tendência a afundar permanece, revelando por que evitar tais intermediários é essencial para heteroestruturas limpas.
Como enxofre extra protege a interface
O grupo então perguntou o que ocorre quando enxofre é introduzido depois que essa camada enterrada se forma. Quando enxofre é adicionado à fase pura SMoMoS, ele pode puxar átomos de molibdênio de volta para a superfície e eventualmente reconstruir uma segunda camada adequada de MoS2 acima da folha original. Entretanto, quando a camada enterrada já é uma liga (SMMS), o enxofre extra puxa tanto átomos de molibdênio quanto de tungstênio para cima, produzindo duas camadas ligadas em vez de uma interface nítida. Simulações adicionais mostraram uma saída: se o molibdênio que chega já estiver ligado ao enxofre — formando clusters Mo–S em vez de átomos metálicos nus — ele deixa de afundar. Em condições ricas em enxofre, esses clusters difundem-se pela superfície, se fundem e reparam defeitos, permitindo que uma segunda camada limpa cresça sem formar a problemática liga enterrada.
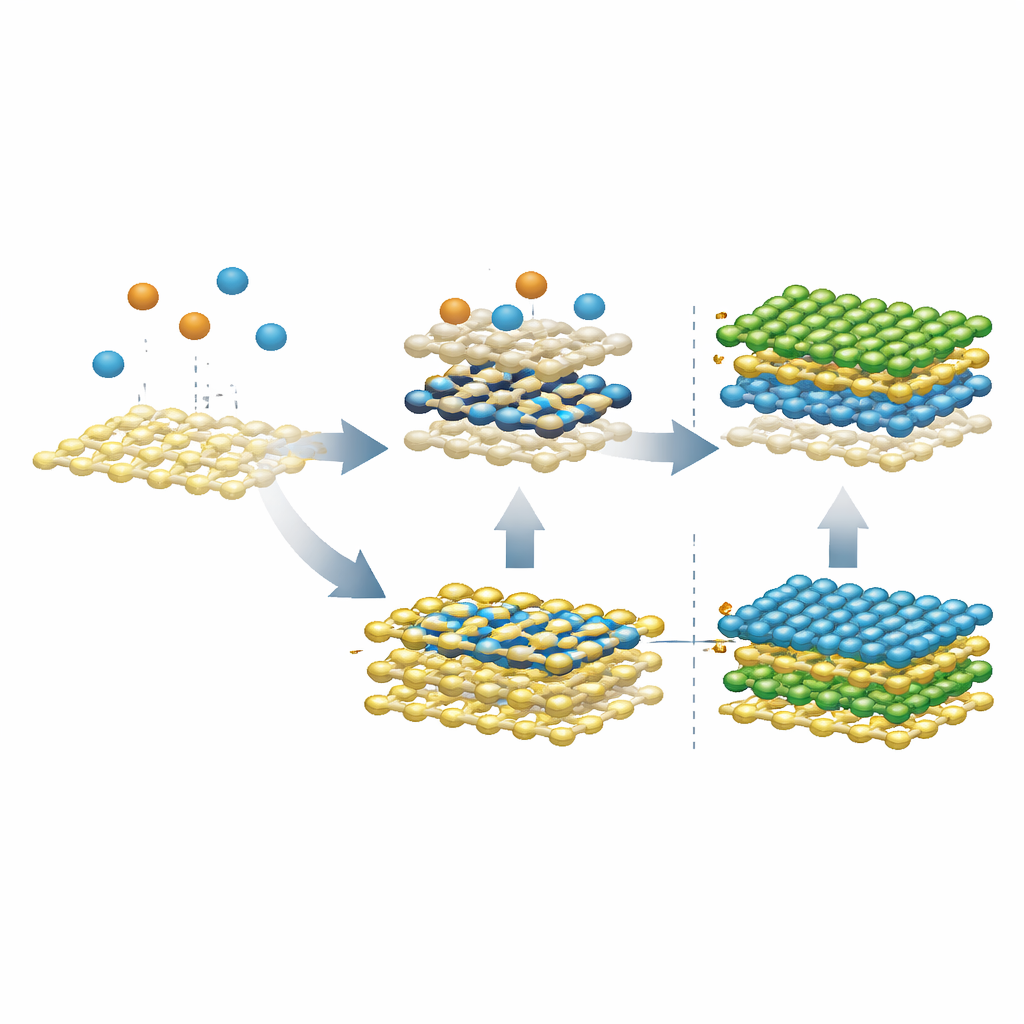
Transformando um problema em um novo tipo de contato
Curiosamente, as mesmas camadas metálicas enterradas que sabotam o empilhamento ordenado podem ser extremamente úteis por si só. Cálculos mostram que SMoMoS e SMMS se comportam como metais e formam contatos tipo p de baixa resistência quando unidos ao MoS2 semicondutor. Ao contrário de muitos eletrodos metálicos convencionais, que sofrem com fortes efeitos de "pinning" que elevam a barreira para buracos, essas interfaces metal–metal coerentes mantêm essa barreira pequena e ajustável. Isso sugere que, se formadas intencionalmente e no local certo, tais camadas intermediárias poderiam servir como eletrodos ideais para transistores ultrafinos.
O que isso significa para a tecnologia atômica
No geral, o estudo revela que o crescimento de materiais empilhados ultrafinos é governado por um equilíbrio delicado entre o afundamento de átomos metálicos nus e a estabilização de clusters ricos em enxofre na superfície. Uma camada metálica enterrada específica, SMMS, emerge como a via-chave para a liga indesejada — mas também como um contato metálico promissor. Para os fabricantes de dispositivos, a mensagem é simples: mantenha condições ricas em enxofre e evite expor camadas existentes a átomos metálicos nus se desejar interfaces nítidas, enquanto crie intencionalmente camadas metálicas enterradas onde contatos de baixa resistência são necessários. Ao transformar um intermediário invisível em um parâmetro de projeto, este trabalho oferece um roteiro tanto para melhor fabricação quanto para um uso mais inteligente dos materiais bidimensionais.
Citação: Zhao, L., Liu, H., Chang, Y. et al. Intermediates of forming transition metal dichalcogenide heterostructures revealed by machine learning simulations. Nat Commun 17, 3086 (2026). https://doi.org/10.1038/s41467-026-69977-x
Palavras-chave: materiais 2D, heteroestruturas van der Waals, simulação com aprendizado de máquina, crescimento de MoS2 WS2, engenharia de contatos