Clear Sky Science · es
Intermedios en la formación de heteroestructuras de dicalcogenuros de metales de transición revelados por simulaciones con aprendizaje automático
Por qué apilar materiales ultrafinos es tan difícil
La electrónica construida a partir de láminas de apenas unos átomos de grosor promete dispositivos más rápidos y eficientes. Una familia popular de estos semiconductores ultrafinos está formada por metales unidos al azufre o al selenio, y cuando se apilan distintas láminas pueden comportarse como materiales completamente nuevos. Pero cultivar pilas grandes y sin defectos en el laboratorio ha resultado complicado: las capas tienden a mezclarse en aleaciones en lugar de permanecer claramente separadas. Este estudio utiliza simulaciones informáticas avanzadas impulsadas por aprendizaje automático para asomarse a los pasos ocultos del crecimiento de tales pilas, revelando una estructura intermedia inesperada que ayuda a explicar tanto los problemas como nuevas oportunidades para los dispositivos.
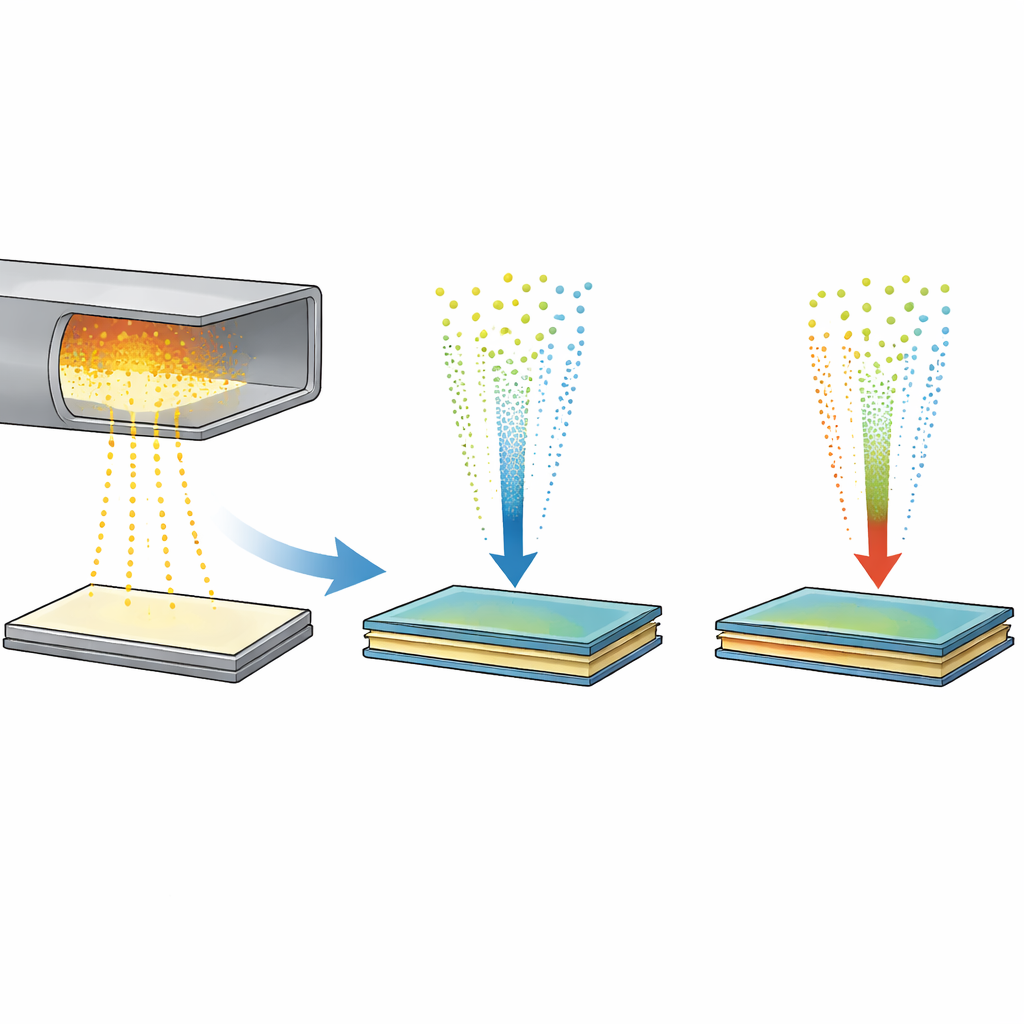
Construir pilas atómicas para los chips del futuro
Los ingenieros están especialmente interesados en apilar materiales como el disulfuro de molibdeno (MoS2) y el disulfuro de tungsteno (WS2). Estas llamadas heteroestructuras de van der Waals conducen bien la electricidad, interactúan fuertemente con la luz y pueden ensamblarse como piezas de Lego a escala atómica. El apilado mecánico puede producir interfaces nítidas y bien definidas, pero solo en pequeñas láminas y a alto coste. Métodos escalables como la deposición química en fase vapor pueden cultivar monocapas en una oblea entera, sin embargo, cuando los investigadores intentan apilar distintas capas, los metales tienden a intercambiar posiciones y formar aleaciones mixtas, estropeando el comportamiento electrónico limpio que requieren los dispositivos.
Usar simulaciones inteligentes como una cámara atómica
Observar el movimiento de los átomos durante el crecimiento en un horno real es casi imposible, por lo que los autores construyeron un modelo digital muy preciso. Entrenaron un potencial basado en aprendizaje automático—un modelo de inteligencia artificial afinado con miles de cálculos cuántico‑mecánicos—para imitar cómo interactúan los átomos de molibdeno, tungsteno y azufre. Integrado en simulaciones de dinámica molecular, este modelo les permitió seguir millones de movimientos atómicos durante nanosegundos manteniendo una precisión cercana a la cuántica. Verificaron que el modelo reproducía fielmente estructuras, energías y vibraciones conocidas, asegurando que sus predicciones sobre las rutas de crecimiento son de confianza.
Una capa metálica enterrada que lo cambia todo
Las simulaciones primero analizaron qué ocurre cuando átomos metálicos desnudos aterrizan sobre una lámina existente de MoS2 o WS2, reflejando un proceso de vapor en dos pasos usado en experimentos. En lugar de permanecer encima como una película ordenada, átomos individuales de molibdeno se hundieron rápidamente en la capa de azufre bajo la superficie, formando una capa metálica enterrada sandwich entre hojas de azufre—etiquetada SMoMoS cuando solo interviene molibdeno, y SMMS cuando se mezclan molibdeno y tungsteno. Esta capa hundida es sorprendentemente estable y fomenta el intercambio de átomos metálicos entre distintas posiciones, lo que conduce de forma natural a la aleación en lugar de a una pila prístina de MoS2/WS2. A temperaturas más bajas el intercambio se ralentiza, pero la tendencia a hundirse persiste, revelando por qué evitar tales intermedios es esencial para heteroestructuras limpias.
Cómo el azufre extra protege la interfaz
El equipo preguntó luego qué ocurre cuando se introduce azufre después de que se forma esta capa enterrada. Cuando se añade azufre a la fase pura SMoMoS, puede arrastrar átomos de molibdeno de vuelta hacia la superficie y reconstruir eventualmente una segunda capa adecuada de MoS2 sobre la lámina original. Sin embargo, cuando la capa enterrada ya es una aleación (SMMS), el azufre adicional tira de ambos, molibdeno y tungsteno, hacia arriba, produciendo dos capas aleadas en lugar de una interfaz nítida. Simulaciones adicionales mostraron una salida: si el molibdeno entrante llega ya ligado al azufre—formando cúmulos Mo–S en lugar de átomos metálicos desnudos—ya no se hunde. En condiciones ricas en azufre, estos cúmulos difunden por la superficie, se fusionan y reparan defectos, permitiendo que crezca una segunda capa limpia sin formar la problemática aleación enterrada.
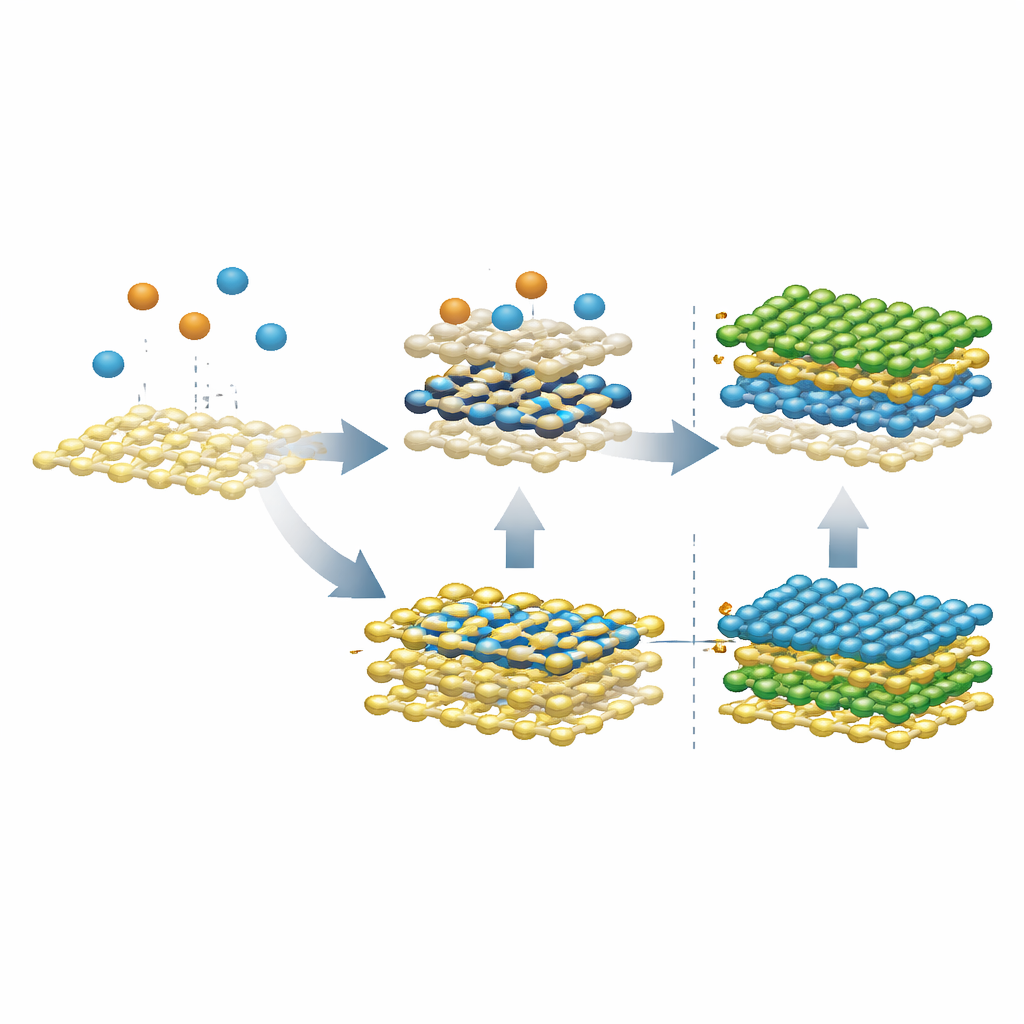
Convertir un problema en un nuevo tipo de contacto
Curiosamente, las mismas capas metálicas enterradas que sabotean el apilado ordenado pueden ser extremadamente útiles por sí mismas. Los cálculos muestran que SMoMoS y SMMS se comportan como metales y forman contactos de tipo p de baja resistencia cuando se unen al MoS2 semiconductivo. A diferencia de muchos electrodos metálicos convencionales, que sufren fuertes efectos de "pinning" que elevan la barrera para huecos, estas interfaces coherentes metal‑metal mantienen esa barrera pequeña y ajustable. Esto sugiere que, si se forman intencionadamente y en el lugar adecuado, tales capas intermedias podrían servir como electrodos ideales para transistores ultrafinos.
Qué significa esto para la tecnología atómica
En conjunto, el estudio revela que el crecimiento de materiales ultrafinos apilados está gobernado por un delicado equilibrio entre el hundimiento de átomos metálicos desnudos y la estabilización de cúmulos ricos en azufre en la superficie. Una capa metálica enterrada específica, SMMS, emerge como la puerta de entrada clave a la aleación no deseada—pero también como un contacto metálico prometedor. Para los fabricantes de dispositivos, el mensaje es simple: mantener condiciones ricas en azufre y evitar exponer capas existentes a átomos metálicos desnudos si se quieren interfaces nítidas, mientras que crear deliberadamente capas metálicas enterradas donde se necesiten contactos de baja resistencia. Al convertir un intermedio invisible en un parámetro de diseño, este trabajo ofrece una hoja de ruta tanto para una mejor fabricación como para un uso más inteligente de materiales bidimensionales.
Cita: Zhao, L., Liu, H., Chang, Y. et al. Intermediates of forming transition metal dichalcogenide heterostructures revealed by machine learning simulations. Nat Commun 17, 3086 (2026). https://doi.org/10.1038/s41467-026-69977-x
Palabras clave: materiales 2D, heteroestructuras de van der Waals, simulación con aprendizaje automático, crecimiento de MoS2 WS2, ingeniería de contactos