Clear Sky Science · pl
Utrata PRC2 upośledza tumorogenezę drobnokomórkowego raka płuca i zwiększa wrażliwość na hamowanie G9a/GLP
Dlaczego te badania mają znaczenie dla osób z rakiem płuca
Drobnokomórkowy rak płuca jest jedną z najbardziej śmiertelnych postaci raka płuca, często szybko się rozsiewa i opiera się dostępnym terapiom, takim jak chemioterapia czy imunoterapia. W tym badaniu postawiono proste, ale istotne pytanie: zamiast ograniczać się wyłącznie do atakowania DNA nowotworu, czy nie dałoby się także przeprogramować przełączników, które mówią komórkom raka, które geny włączać lub wyłączać? Dzięki temu badacze odkryli nową słabość drobnokomórkowego raka płuca, która może zwiększyć skuteczność istniejących leków i otworzyć drogę do bardziej przemyślanych terapii skojarzonych.
Śmiertelny nowotwór ukrywający się przed układem odpornościowym
Drobnokomórkowy rak płuca jest zwykle rozpoznawany późno, a pięcioletnie przeżycie wynosi poniżej 7 procent. Wiele guzów zachowuje tożsamość „neuroendokrynną”, co oznacza, że przypominają komórki nerwowe produkujące hormony. Guzy te mają tendencję do wzrostu w postaci luźnych, unoszących się skupisk, ukrywają się przed układem odpornościowym i szybko nabywają oporność na chemioterapię. Tylko niewielka część pacjentów ma guzy zapalne i rozpoznawalne przez komórki odpornościowe, dlatego leki przeciwko punktom kontrolnym pomagają tak niewielu osobom. Z tego powodu naukowcy zwracają uwagę na epigenom nowotworu — chemiczne znaczniki na DNA i białkach je pakujących, które działają jak oprogramowanie genomu — by sprawdzić, czy zresetowanie tych znaczników może uczynić guzy mniej agresywnymi i łatwiejszymi do zniszczenia.
Mistrzowski kompleks wyciszający geny o dwóch obliczach
Badanie koncentruje się na maszynie białkowej zwanej PRC2, która normalnie wyłącza zestawy genów poprzez nakładanie chemicznych znaczników na białka, wokół których owinięte jest DNA. PRC2 ma dwa kluczowe aspekty: strukturę fizyczną, która utrzymuje kompleks w całości, oraz aktywność enzymatyczną, która faktycznie dodaje znaki wyciszające. Używając mysich modeli zaprojektowanych do rozwoju drobnokomórkowego raka płuca, badacze usunęli podstawowy element strukturalny PRC2 zwany EED tuż na początku formowania się guza. Bez tego składnika oczekiwane guzy płuc prawie wcale się nie tworzyły, a rzadkie zmiany, które się pojawiły, rosły słabo i pozbawione były typowych cech neuroendokrynnych. Komórki nowotworowe tracące EED obumierały, a nawet lek zaprojektowany do rozkładu EED selektywnie zabijał zarówno mysie, jak i ludzkie komórki drobnokomórkowego raka płuca, oszczędzając komórki gruczolakoraka płuca. Pokazało to, że integralność strukturalna PRC2 jest niezbędna zarówno do zapoczątkowania, jak i utrzymania tego nowotworu.
Samym blokowaniem enzymu nie da się wystarczająco osiągnąć
Zespół zapytał następnie, czy leki blokujące aktywność enzymatyczną PRC2 — konkretnie inhibitory EZH2, które są już zatwierdzone dla niektórych nowotworów krwi — mogą naśladować efekt usunięcia EED. W hodowlach komórek drobnokomórkowego raka płuca te leki skutecznie usuwały chemiczne markery nakładane przez PRC2, ale przez kilka dni miały tylko skromny wpływ na przeżycie komórek. W przeszczepach nowotworów u myszy długotrwałe leczenie obniżało te markery w guzach, lecz nie przedłużało życia zwierząt. Jednak gdy leki podawano przez dłuższy czas w warunkach hodowlanych, wywoływały znacznie szersze zmiany w aktywności genów, w tym zwiększone wyrażanie genów związanych z markerami powierzchni komórkowej i rozpoznawaniem przez układ odpornościowy. Komórki nowotworowe zaczęły też tracić cechy neuroendokrynne i przybierać bardziej przylegający, mniej unoszący się styl wzrostu, co sugeruje, że przedłużone hamowanie enzymu stopniowo przesuwa komórki w kierunku innego, bardziej podatnego stanu, zamiast je od razu zabijać.
Odkrycie lekoopornej słabości
Aby wykorzystać ten nowy stan, badacze przeprowadzili ukierunkowane badanie łączące inhibitory EZH2 z panelem innych leków. Odkryli, że po wstępnym leczeniu inhibitorem EZH2 neuroendokrynne komórki drobnokomórkowego raka płuca stały się wysoce wrażliwe na blokowanie innej pary enzymów, G9a i GLP, które również kontrolują wyciszanie genów przez inne chemiczne znaczniki. Ta nadwrażliwość występowała tylko w liniach neuroendokrynnych drobnokomórkowego raka płuca, a nie w powiązanych liniach nieneuroendokrynnych ani w komórkach międzybłoniaka. U myszy niosących ludzkie przeszczepy drobnokomórkowego raka płuca sam inhibitor G9a/GLP dawał umiarkowaną korzyść w przeżyciu, inhibitor EZH2 sam w sobie nie pomagał, ale kombinacja znacząco wydłużała przeżycie. Analizy molekularne wykazały, że hamowanie EZH2 przeorganizowuje sieć białek współdziałających z PRC2 i wraz z hamowaniem G9a/GLP silnie zwiększa ekspresję genów zaangażowanych w stres oksydacyjny i oksydazy komórkowe, w tym enzymy napędzające utlenianie lipidów.
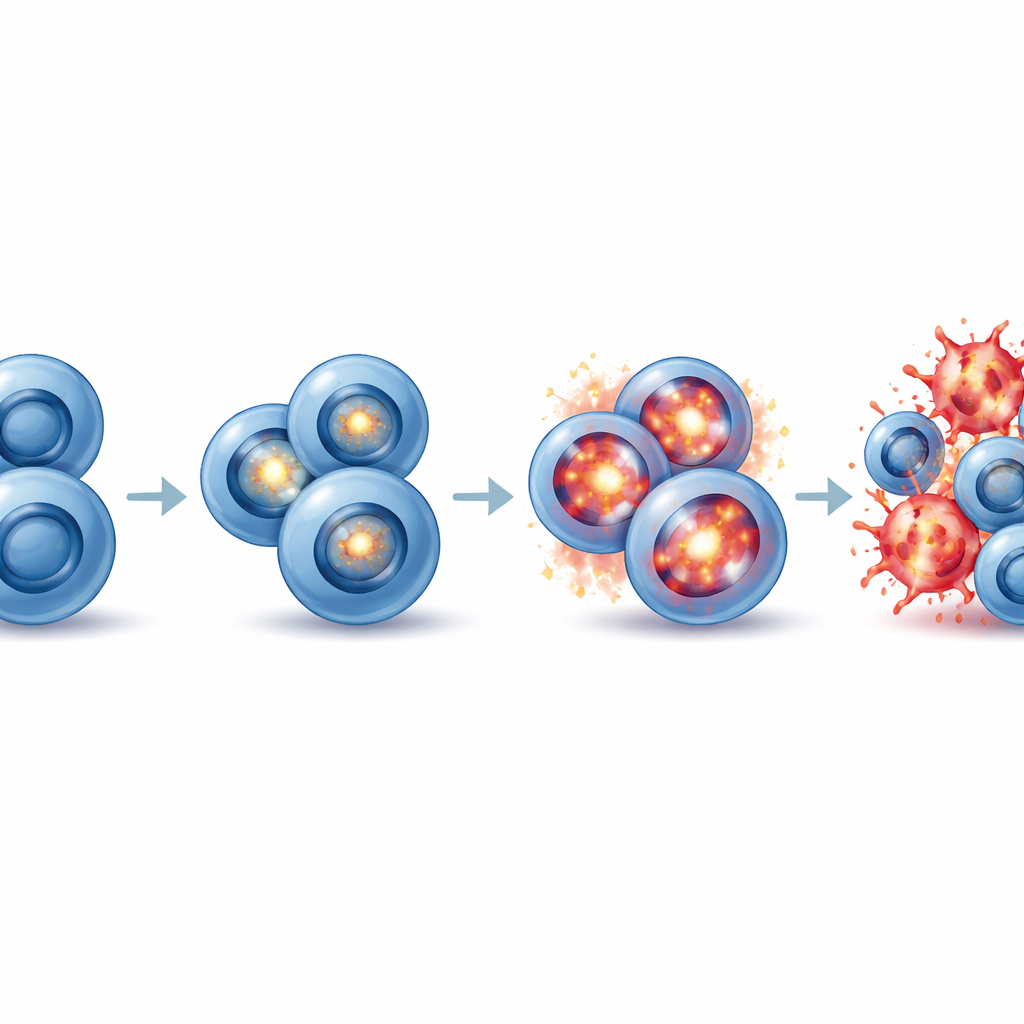
Wypchnięcie komórek nowotworowych w śmiertelne przeciążenie oksydacyjne
Stres oksydacyjny pojawia się, gdy reaktywne cząsteczki tlenu gromadzą się szybciej, niż komórki są w stanie je neutralizować, uszkadzając tłuszcze, białka i DNA. Badanie wykazało, że połączenie hamowania EZH2 i G9a/GLP wywołuje gwałtowny wzrost stresu oksydacyjnego specyficznie w neuroendokrynnych komórkach drobnokomórkowego raka płuca. Zwiększyły się markery uszkodzeń oksydacyjnych, a bezpośrednie pomiary przy użyciu fluorescencyjnego wskaźnika potwierdziły wyższe poziomy reaktywnych form tlenu tylko w komórkach neuroendokrynnych traktowanych kombinacją. Gdy badacze dodali powszechny antyoksydant, N-acetyl-L-cysteinę, uratował on komórki przed śmiercią, dowodząc, że stres oksydacyjny był główną przyczyną ich zagłady. Komórki nieneuroendokrynne nie doświadczyły tego skoku, co tłumaczy, dlaczego były oszczędzone. W istocie, przedłużone hamowanie EZH2 „primuje” komórki neuroendokrynne drobnokomórkowego raka płuca tak, że kolejny cios w G9a/GLP przeciąża ich mechanizmy obronne przed stresem i popycha je za oksydacyjny klif.
Co to znaczy dla przyszłych terapii
Dla laika kluczowa wiadomość jest taka: to badanie rozdziela dwie role tej samej maszyny wyciszającej geny w drobnokomórkowym raku płuca. Strukturalne jądro PRC2, w tym EED, jest absolutnie wymagane, by ten nowotwór w ogóle istniał, co czyni je atrakcyjnym celem dla nowych leków-degradujących, które fizycznie rozbierają kompleks. Natomiast samo wyłączenie części enzymatycznej istniejącymi inhibitorami nie zatrzymuje guzów samodzielnie, ale przekształca komórki nowotworowe w stan, który odsłania ukrytą słabość: zależność od precyzyjnie kontrolowanego stresu oksydacyjnego. Łącząc przedłużone hamowanie EZH2 z inhibitorami G9a/GLP, badacze byli w stanie wykorzystać tę słabość i selektywnie zabić trudne do leczenia guzy neuroendokrynne przez przeciążenie oksydacyjne. Koncepcja najpierw „popychania” komórek nowotworowych w bardziej podatną tożsamość, a następnie uderzania w ich nową piętę Achillesa, może pomóc zaprojektować mądrzejsze, trwalsze strategie terapeutyczne dla pacjentów z tą agresywną chorobą.
Cytowanie: Kopparam, J., Chandrasekaran, G., Hulsman, D. et al. PRC2 loss impairs small cell lung cancer tumorigenesis and enhances sensitivity to G9a/GLP inhibition. Commun Biol 9, 469 (2026). https://doi.org/10.1038/s42003-026-09677-w
Słowa kluczowe: drobnokomórkowy rak płuca, terapia epigenetyczna, PRC2, inhibitor EZH2, stres oksydacyjny