Clear Sky Science · pl
Postępujące upośledzenie poznawcze i tachykardia komorowa u chłopca z biallelicznymi wariantami POLG oraz de novo wariacją RYR2
Kiedy dwa genetyczne defekty się spotykają
Większość z nas myśli o chorobie jako o jednym jej przyczynie, ale lekarze odkrywają, że niektórzy pacjenci chorują z powodu więcej niż jednej ukrytej wady w DNA. Ten artykuł opowiada historię chłopca, którego pogarszające się trudności w nauce i nagły, śmiertelny zaburzenie rytmu serca ostatecznie przypisano dwóm odrębnym uszkodzeniom genetycznym — jedno wpływało na maleńkie elektrownie w komórkach, a drugie zaburzało elektryczny rytm serca. Jego przypadek pokazuje, jak nowoczesne narzędzia genetyczne i sprytne modele laboratoryjne, w tym komórki drożdżowe, potrafią rozplątać takie medyczne zagadki i poprawić diagnostykę dla przyszłych pacjentów.
Zagadkowa historia mózgu i serca
Chłopiec z tego raportu rozwijał się początkowo prawidłowo, poza opóźnioną mową. W wieku pięciu lat jego zdolności poznawcze mieściły się w normie, lecz w ciągu następnych kilku lat jego wyniki szkolne gwałtownie się pogorszyły i do dziewiątego roku życia spełniał kryteria niepełnosprawności intelektualnej. Badania obrazowe mózgu i wczesne testy serca nie wykazywały istotnych odchyleń, a standardowe badania genetyczne nie ujawniły przyczyny. Potem, w wieku dwunastu lat, zaczął mieć omdlenia związane z nieprawidłowymi szybkimi rytmami w górnych jamach serca. Pomimo leczenia farmakologicznego funkcja serca pogarszała się i zmarł nagle z powodu zatrzymania krążenia w wieku trzynastu lat. Połączenie postępujących problemów poznawczych i niebezpiecznych zaburzeń rytmu serca skłoniło do głębszego przeszukania jego DNA.
Znajdowanie dwóch odrębnych winowajców genetycznych
Przy użyciu sekwencjonowania następnej generacji, które potrafi przeskanować setki genów jednocześnie, zespół najpierw znalazł trzy zmiany w genie o nazwie POLG. Gen ten koduje kluczową część enzymu kopiującego DNA mitochondrialne — małego pierścienia materiału genetycznego wewnątrz „elektrowni” komórki. Dwie zmiany tworzyły znaną szkodliwą parę na chromosomie ojca, wcześniej powiązaną z chorobą mitochondrialną. Trzecia zmiana, nazwana W113R, pochodziła od matki i nigdy wcześniej nie została opisana, co utrudniało ocenę jej znaczenia klinicznego. Później, bardziej ukierunkowana ponowna analiza danych egzomu chłopca wykryła dodatkową zmianę w innym genie, RYR2, który pomaga kontrolować przepływ wapnia w komórkach mięśnia sercowego. Ten wariant RYR2, Y4725C, był opisywany u innych pacjentów z zagrażającymi życiu szybki rytmami serca i nie występował u żadnego z rodziców, co oznacza, że pojawił się de novo u chłopca.
Wykorzystanie drożdży do przetestowania nowej mutacji
Aby ustalić, czy nieznany wariant POLG rzeczywiście uszkadza enzym, badacze zwrócili się do nieoczekiwanego sprzymierzeńca: drożdży piekarskich. Komórki drożdży mają własną wersję tej samej polimerazy mitochondrialnej, nazwaną Mip1. Ponieważ dotknięty aminokwas jest zachowany między gatunkami, zespół wprowadził ludzką zmianę W113R do genu drożdżowego w odpowiadającej pozycji. Następnie porównano drożdże z normalnym Mip1 z drożdżami noszącymi zmienioną wersję. W normalnej temperaturze mutantyczne drożdże potrafiły rosnąć oddychając tlenowo, ale wykazywały więcej komórek, które utraciły lub uszkodziły swoje DNA mitochondrialne. Pod wpływem stresu cieplnego te problemy stały się dramatyczne: niemal wszystkie komórki z mutantycznym enzymem nie miały zdolności do oddychania, a zawartość ich DNA mitochondrialnego gwałtownie spadła. Mutantyczny enzym powodował też nieco więcej błędów podczas kopiowania, mimo że poziom samego białka nie był zmniejszony. Razem wyniki te wykazały, że zmiana W113R upośledza utrzymanie DNA mitochondrialnego, szczególnie pod wpływem stresu.
Skomponowanie całości obrazu
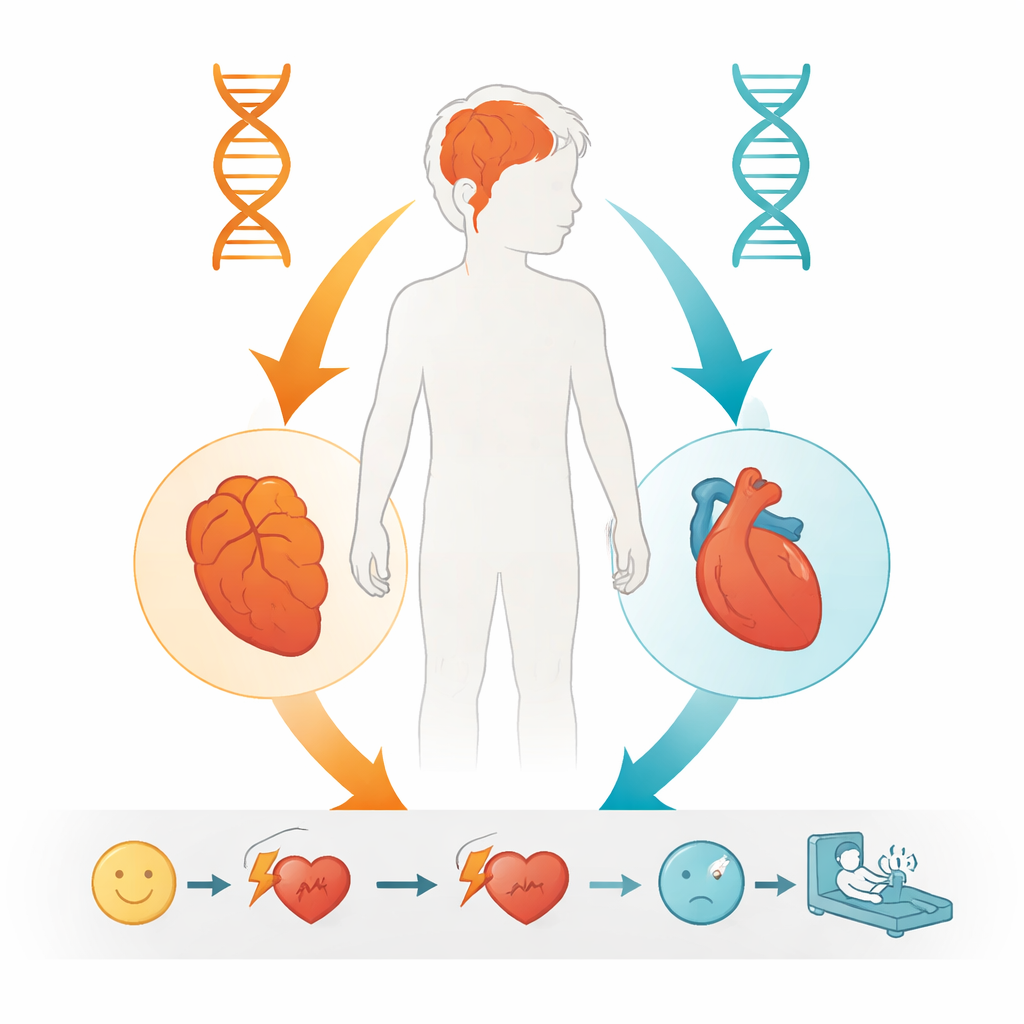
Mając te ustalenia, autorzy doszli do wniosku, że pogarszające się problemy poznawcze chłopca najlepiej tłumaczyły jego „podwójne uderzenie” w POLG: jedna znana szkodliwa allela od ojca i nowo udowodniona szkodliwa allela W113R od matki. Pasuje to do opisów innych pacjentów mających dwie wadliwe kopie POLG i rozwijających postępujące objawy neurologiczne zaczynające się w dzieciństwie lub młodzieńczości. Z kolei zaburzenie rytmu serca bardzo przypominało to obserwowane u osób z wariantem Y4725C w RYR2, genie dobrze znanym z wywoływania niebezpiecznej tachykardii komorowej. Choć autorzy nie mogą całkowicie wykluczyć pewnej interakcji między defektami mitochondrialnymi a zaburzeniem kanału sercowego, najczytelniejszy obraz wskazuje, że dwie w dużej mierze niezależne wady genetyczne wpłynęły równolegle na mózg i serce chłopca.
Dlaczego ten przypadek ma znaczenie
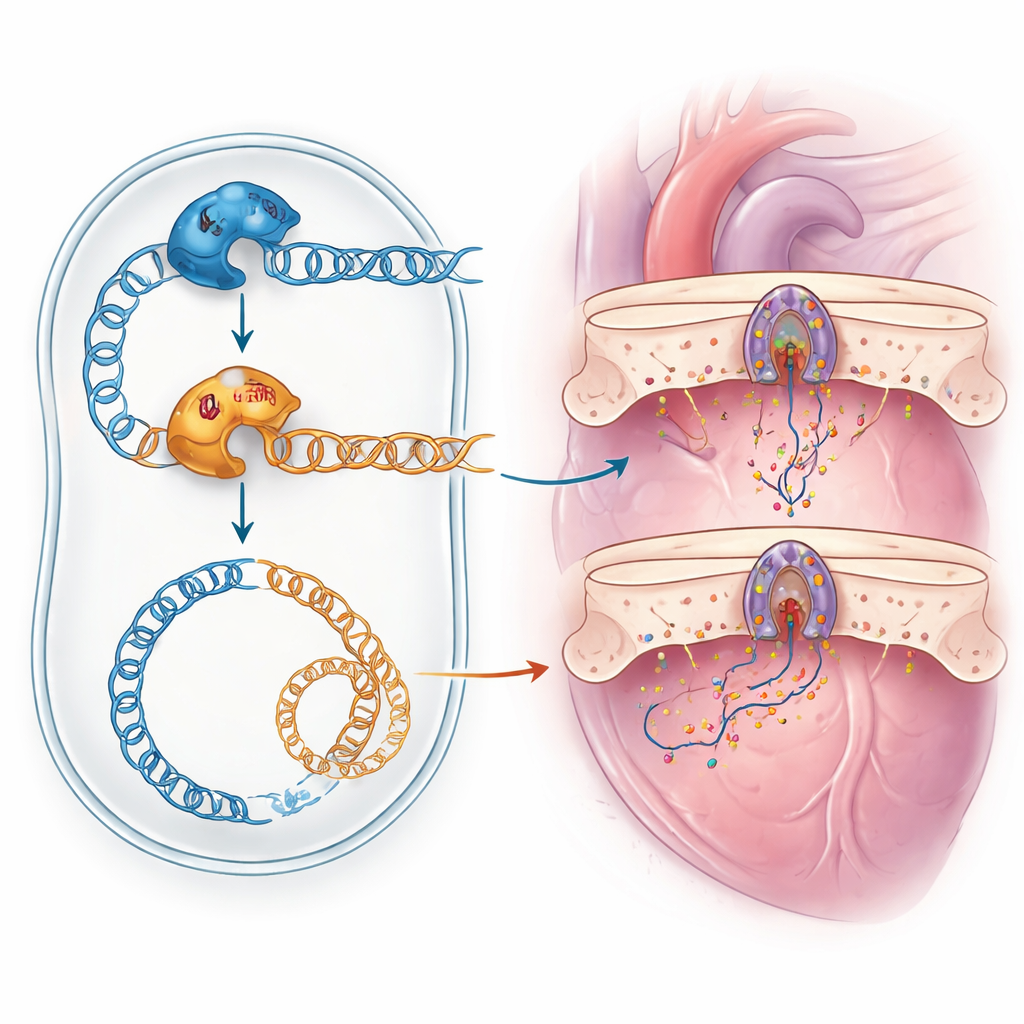
Ten przypadek podkreśla kilka lekcji dla współczesnej medycyny. Po pierwsze, złożone objawy niektórych pacjentów nie wynikają z jednej rzadkiej choroby, lecz z „podwójnego kłopotu” spowodowanego więcej niż jedną wadą genetyczną, co łatwo przeoczyć bez starannej ponownej analizy danych sekwencyjnych. Po drugie, same programy komputerowe nie zawsze potrafią wiarygodnie ocenić, czy nowo odkryta zmiana DNA jest szkodliwa; testy funkcjonalne, takie jak modelowanie mutacji w drożdżach, mogą dostarczyć rozstrzygających dowodów. Wreszcie, wyjaśnienie, który gen odpowiada za które objawy, jest kluczowe dla dokładnego poradnictwa rodzinom oraz dla dopasowania pacjentów do pojawiających się terapii, na przykład eksperymentalnych leków mających na celu ratowanie defektów mitochondrialnych związanych z POLG. Dla rodzin i klinicystów stających wobec niewyjaśnionych, wieloukładowych chorób, to badanie pokazuje, jak połączenie szerokich badań genetycznych z inteligentnymi modelami laboratoryjnymi w końcu może dostarczyć odpowiedzi.
Cytowanie: Fumini, V., Gilea, A.I., Tacchetto, E. et al. Progressive cognitive impairment and ventricular tachycardia in a boy with biallelic POLG variants and a de novo RYR2 variation. Sci Rep 16, 14289 (2026). https://doi.org/10.1038/s41598-026-44913-7
Słowa kluczowe: choroba mitochondrialna, genetyczna arytmia, mutacja POLG, kanalopatia RYR2, funkcjonalny test w drożdżach