Clear Sky Science · pl
Blokada PD-1 wraz z inhibitorem kinazy tyrozynowej przebudowuje mikrośrodowisko guza w zaawansowanym raku nerki
Dlaczego leczenie raka nerki może przestać działać
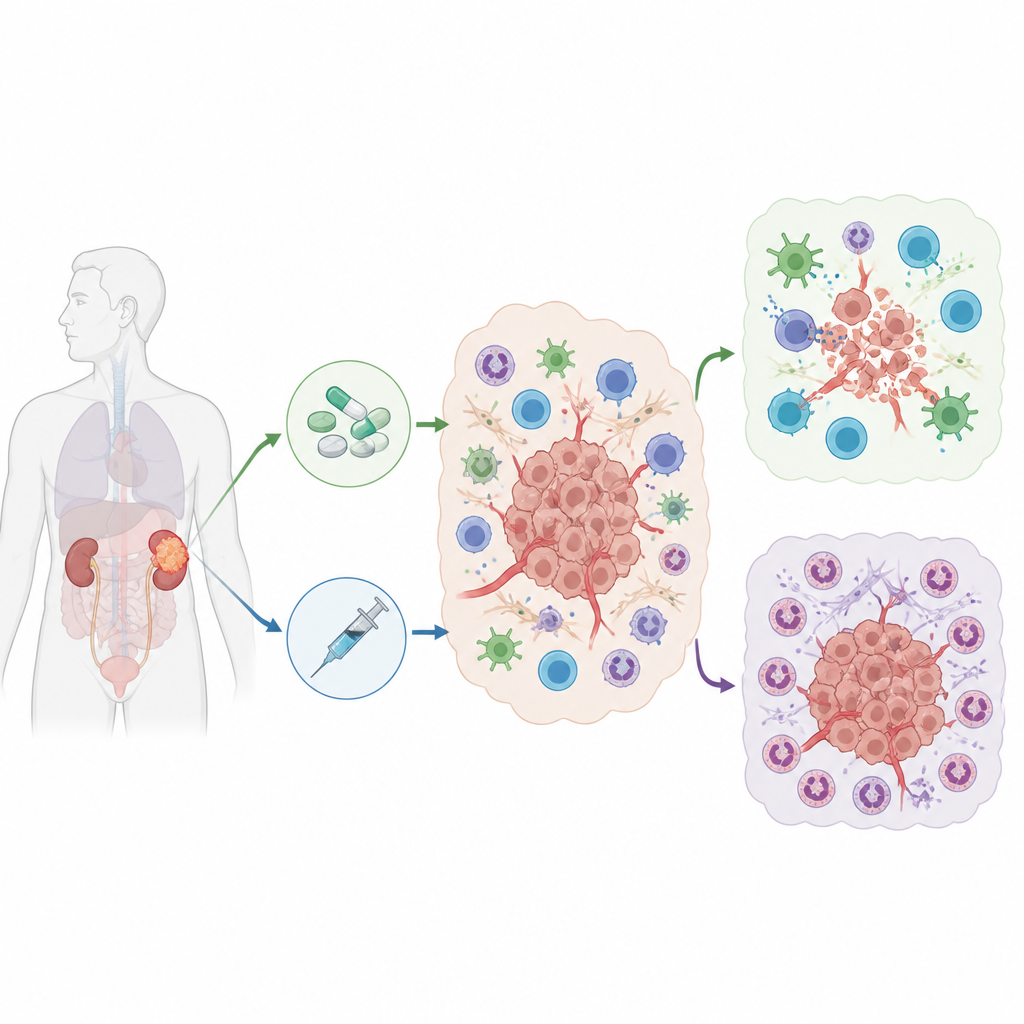
Wiele osób z zaawansowanym rakiem nerki otrzymuje obecnie silne połączenie leków: jeden preparat ogranicza dopływ krwi do guza, a drugi wzmacnia odpowiedź układu odpornościowego. Mimo to niepokojąca liczba pacjentów przestaje reagować na terapię. W tym badaniu zadano proste, lecz kluczowe pytanie: co w obrębie guza sprawia, że niektórzy pacjenci korzystają z tej kombinacji, podczas gdy inni doświadczają nawrotu albo w ogóle nie reagują?
Sąsiedztwo wokół guza
Guzy nie rozwijają się w izolacji; żyją w zatłoczonym otoczeniu komórek odpornościowych, naczyń krwionośnych i tkanki podporowej, znanym jako mikrośrodowisko guza. Badacze zebrali 61 próbek guza od 34 osób z zaawansowanym rakiem nerki, które były leczone lekami hamującymi naczynia, lekami immunologicznymi lub obiema grupami leków. Przy użyciu sekwencjonowania RNA pojedynczych komórek, techniki odczytującej aktywność tysięcy genów w pojedynczych komórkach, zbudowali szczegółową mapę niemal 333 000 komórek. Mapa ujawniła wiele typów komórek, w tym komórki nowotworowe, limfocyty T zdolne zabijać guzy oraz kilka rodzajów białych krwinek, które mogą wspierać lub hamować odpowiedź immunologiczną.
Komórki odpornościowe pomocne i szkodliwe
Porównując pacjentów reagujących na leczenie z tymi, którzy nie reagowali, zespół odkrył wyraźne wzorce. Osoby, które odpowiadały na terapię, miały zwykle więcej limfocytów T wewnątrz guza i mniej komórek z grupy mieloidalnej, obejmującej makrofagi i neutrofile. U pacjentów niereagujących obserwowano odwrotny obraz, szczególnie po terapii skojarzonej. Dokładniejsza analiza neutrofili — typu szybko reagujących białych krwinek — ujawniła kilka odrębnych podgrup. Jedna podgrupa, charakteryzująca się silnymi sygnałami związanymi z angiogenezą i metabolizmem beztlenowym, znacznie się rozrosła u niereagujących po leczeniu. Te neutrofile wiązały się z gorszymi wynikami i oznakami tłumienia aktywności limfocytów T.
Sygnał z komórek nowotworowych przyciągający tłumiące komórki
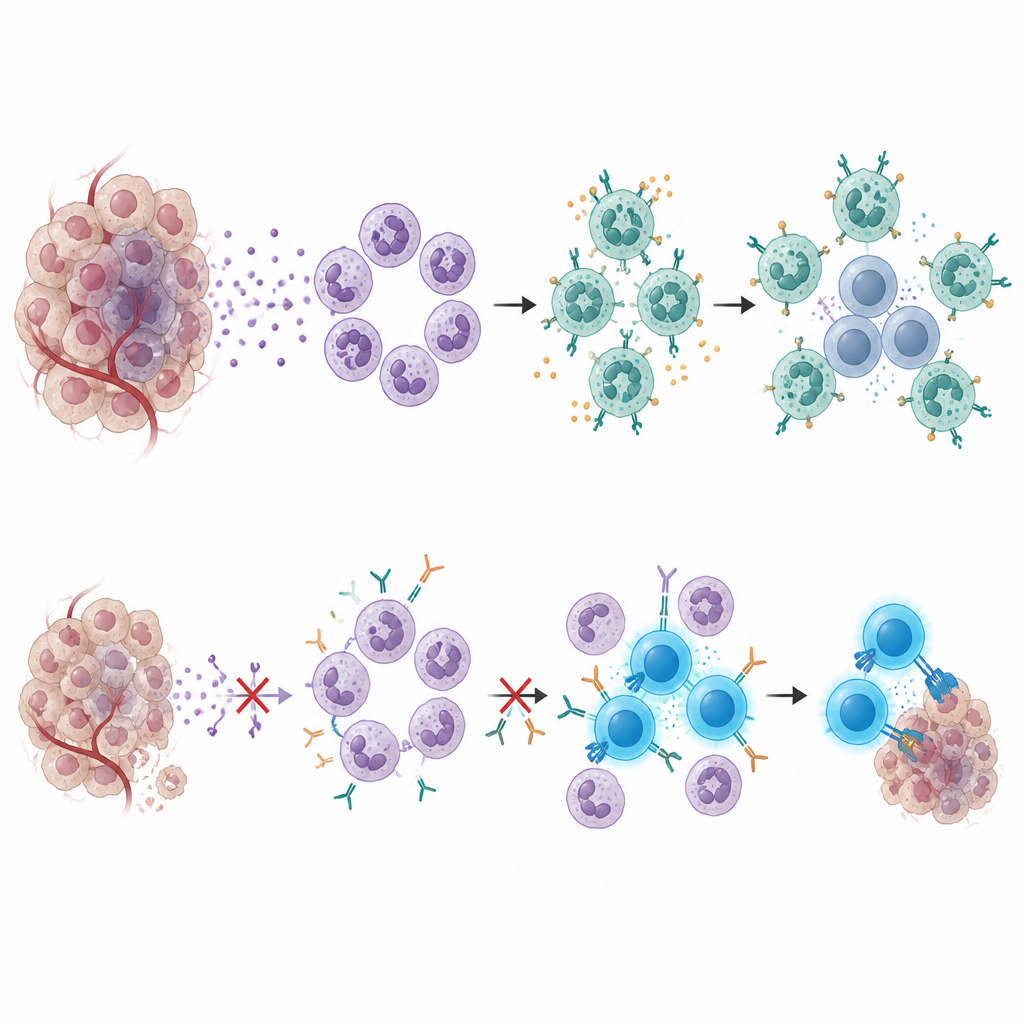
Same komórki nowotworowe odegrały kluczową rolę w przyciąganiu tych szkodliwych neutrofili. W guzach pacjentów niereagujących komórki nowotworowe włączały geny związane z zapaleniem i rekrutacją neutrofili. Dwa z tych genów kodują białko nazywane surowiczym białkiem amyloidowym (SAA), cząsteczkę zwykle obserwowaną przy silnych reakcjach zapalnych. Guzy obfitujące w komórki wyrażające SAA były częstsze u pacjentów opornych na terapię, a wyższe poziomy SAA we krwi przed leczeniem korelowały z gorszym przeżyciem. W wielu zestawach danych z różnych nowotworów pacjenci z wysokim poziomem SAA mieli tendencję do gorszych efektów po terapiach opartych na immunoterapii, co sugeruje, że SAA oznacza uporczywy, oporny stan guza.
Obieg komunikacyjny, który wyłącza limfocyty T
Łącząc mapy komórek, dane przestrzenne i eksperymenty na myszach, naukowcy zrekonstruowali obieg komunikacyjny. SAA uwalniane przez komórki nowotworowe popycha neutrofile w kierunku stanu supresyjnego, w którym produkują VEGFA i białko powierzchniowe zwane CEACAM1. Te neutrofile często gromadzą się blisko podzbioru limfocytów CD8, które wykazują inny białkowy punkt kontrolny — TIM-3. Sygnały przekazywane z CEACAM1 na neutrofilach do TIM-3 na limfocytach T prowadzą do ich wyczerpania, pozostawiając je niezdolne do eliminacji komórek nowotworowych nawet w obecności leków blokujących PD-1. U myszy blokada SAA zwiększała wrażliwość guzów na kombinację leków i zmniejszała nagromadzenie neutrofili, natomiast zahamowanie interakcji CEACAM1–TIM-3, szczególnie razem z inhibitorem PD-1, przywracało aktywność komórek T i spowalniało wzrost guza.
Nowe cele, by przedłużyć skuteczność terapii
Dla osoby niebędącej specjalistą przesłanie tego badania jest takie: niektóre guzy nerek wymykają się współczesnej terapii skojarzonej, budując ochronną osłonę z wyspecjalizowanych neutrofili i wyczerpanych limfocytów T. Białko wytwarzane przez guz, SAA, przyciąga i kształtuje te neutrofile, które następnie uciszają pobliskie limfocyty T poprzez hamujący układ CEACAM1–TIM-3. Pomiar SAA we krwi mógłby pomóc wyłapać pacjentów o wysokim ryzyku oporności, a leki blokujące SAA lub połączenie CEACAM1–TIM-3 mogą w przyszłości zostać dodane do obecnych schematów leczenia. Zakłócając tę ukrytą drogę ucieczki immunologicznej, przyszłe terapie mogłyby zapewnić większej liczbie pacjentów dłuższe korzyści z immunoterapii raka nerki.
Cytowanie: Gu, L., Zhang, Q., Liang, Q. et al. PD-1 blockade plus tyrosine kinase inhibitor remodels the tumor microenvironment in advanced renal cell carcinoma. Nat Commun 17, 4626 (2026). https://doi.org/10.1038/s41467-026-70978-z
Słowa kluczowe: rak nerkowokomórkowy, mikrośrodowisko guza, neutrofile, terapia hamująca punkty kontrolne immunologiczne, oporność na leczenie