Clear Sky Science · pl
Mutacja missense w Kdf1 powoduje defekty szkliwa przez zaburzenie adhezji komórek i sygnalizacji Hippo-YAP w nabłonku zębowym
Dlaczego małe białko zęba ma znaczenie
Szkliwo to błyszcząca biała zbroja, która chroni nasze zęby przed całym życiem przeżuwania, gorącą kawą i zimnymi lodami. U niektórych rodzin ta zbroja nigdy się nie formuje w pełni, przez co zęby są kruche, nadwrażliwe i bardziej podatne na próchnicę — stan znany jako amelogenesis imperfecta. To badanie prowadzi do źródła problemu: subtelnej zmiany w jednym białku o nazwie KDF1 w komórkach tworzących szkliwo, ukazując, jak drobna zmiana genetyczna może osłabić zarówno współpracę komórek, jak i sygnały kierujące rozwojem zęba.
Od solidnej tarczy do kruchej powłoki
Szkliwo wytwarzają wyspecjalizowane komórki powierzchniowe zwane ameloblastami, które precyzyjnie wydzielają i utwardzają białkową warstwę na rosnących zębach. Badacze przeanalizowali mutację w genie KDF1 pochodzącą od pacjenta, powiązaną z brakującymi zębami i słabym szkliwem. U myszy zmodyfikowanych tak, aby nosiły tę samą mutację, stwierdzono, że zarówno osobniki z jedną kopią mutacji (heterozygotyczne), jak i z dwiema kopiami (homozygotyczne) rozwijały cieńsze szkliwo o zmniejszonej zawartości minerałów i nieuporządkowanej strukturze wewnętrznej. Obrazowanie wykazało mniejsze korony zębów, mniejszą objętość szkliwa i niższą gęstość szkliwa, szczególnie u zwierząt z dwiema kopiam i mutacji.
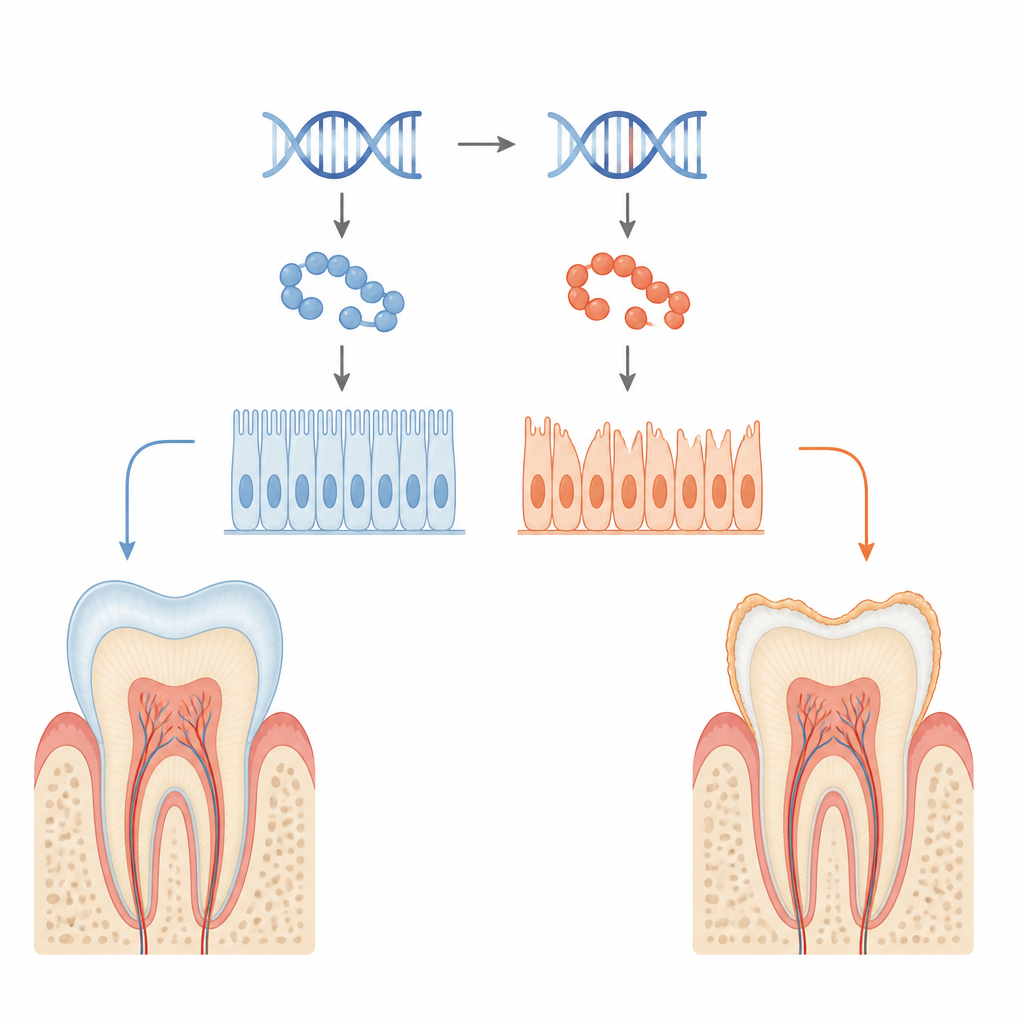
Kiedy komórki budujące zęby tracą przyczepność
Badając powierzchnię zęba bliżej, zespół zaobserwował, że ameloblasty z mutacją nie przylegały mocno do siebie ani do formującej się warstwy szkliwa. Mikroskopia ujawniła szczeliny tam, gdzie komórki powinny być ściśle przymocowane, a obrazy ultrastrukturalne pokazały skrócone i rozciągnięte normalne pasmowe połączenia między sąsiednimi komórkami. Kluczowe cząsteczki adhezyjne działające jak biologiczne nity, takie jak E-kadheryna i integryna β4, były wyraźnie zredukowane u zwierząt z mutacją. W hodowlach komórkowych linii tworzących szkliwo, genetycznie zmodyfikowanych tak, by wytwarzały mutant KDF1, komórki mniej się przyczepiały, bardziej migrowały i wykazywały większą proliferację, ale słabsze cechy dojrzewania, odzwierciedlając defekty zaobserwowane w tkance.
Sygnalizacyjne światła drogowe wyrwane z równowagi
Adhezja komórek to nie tylko mechanika; wpływa też na wewnętrzne sieci sygnałowe, które mówią komórkom, kiedy mają rosnąć, a kiedy się wyspecjalizować. Sekwencjonowanie RNA ameloblastów z myszy z mutacją wykazało szerokie zmiany w genach związanych z macierzą zewnątrzkomórkową, adhezją i ścieżką znaną jako Hippo-YAP, która kontroluje wielkość narządu i los komórek. W zdrowych zębach ta ścieżka utrzymuje białko YAP przeważnie nieaktywnym w cytoplazmie, ograniczając sygnały wzrostowe. U mutantów YAP był mniej fosforylowany, gromadził się w jądrze i tworzył więcej kompleksów ze swoim partnerem TEAD1, włączając geny związane ze wzrostem. Ten wzorzec odpowiadał zaobserwowanemu wzrostowi podziałów komórkowych w obszarach, które normalnie zwalniają, gdy komórki szkliwa dojrzewają.
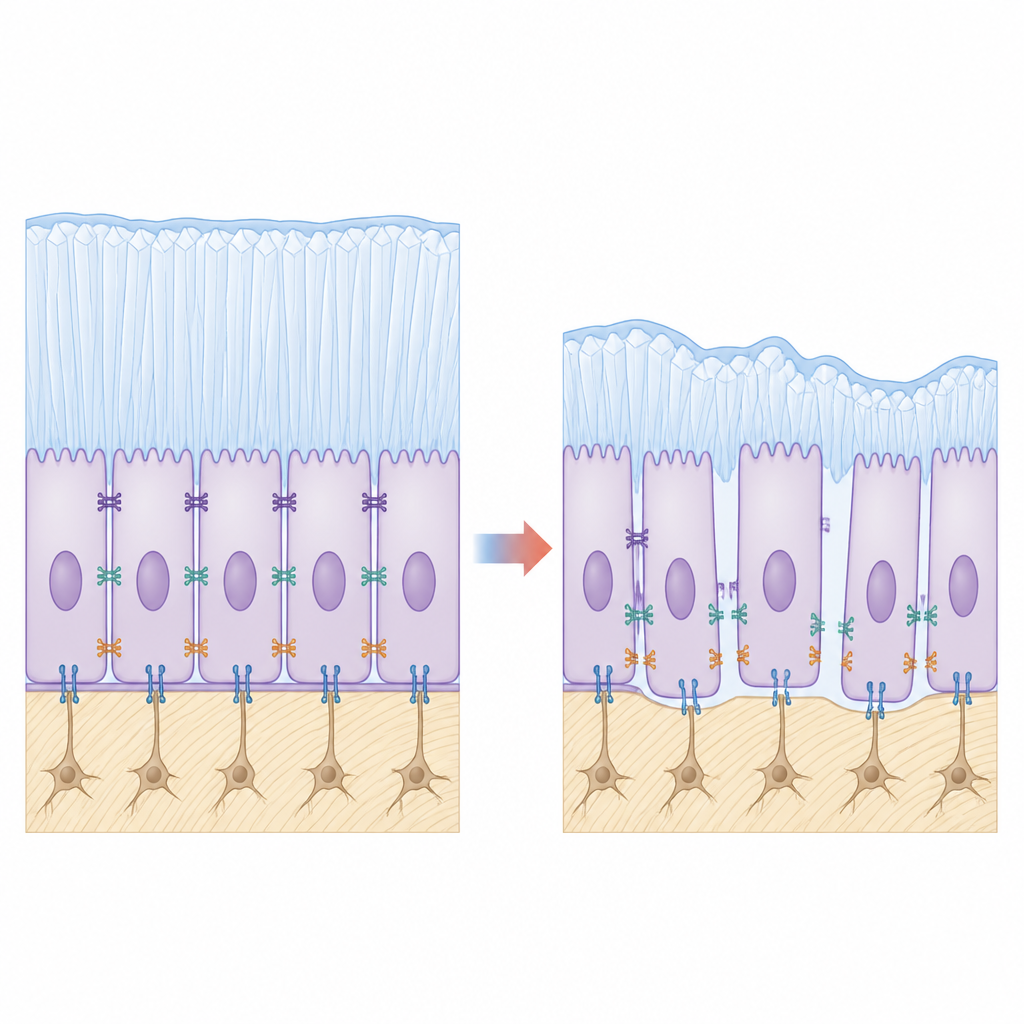
Łączenie osłabionych połączeń z uszkodzonymi sygnałami
Aby powiązać te obserwacje, zespół eksperymentalnie obniżył lub zwiększył poziomy białek adhezyjnych w modelach komórkowych. Redukcja E-kadheryny lub integryny β4 w normalnych komórkach osłabiła aktywność Hippo-YAP w sposób podobny do mutacji KDF1, podczas gdy przywrócenie tych białek adhezyjnych w komórkach mutantów częściowo odnowiło fosforylację YAP, przesuwając układ z powrotem w stronę równowagi. Wspiera to model, w którym błonowo związane KDF1 stabilizuje kompleksy adhezyjne; kiedy KDF1 jest zmutowane i błędnie zlokalizowane, te kompleksy się rozpadają, hamulce Hippo-YAP wysuwają się, komórki nadal się dzielą i nie przechodzą w pełni w stan wyspecjalizowany do wytwarzania szkliwa.
Wskazówka dotycząca przyszłej ścieżki leczenia
Naukowcy przetestowali także verteporfinę, istniejący lek zakłócający partnerstwo YAP-TEAD1. W hodowlach komórek tworzących szkliwo verteporfina zmniejszyła nadmierną proliferację i poprawiła markery produkcji macierzy szkliwa. U młodych myszy z mutacją wczesne leczenie verteporfiną zwiększyło objętość szkliwa, choć nie przywróciło w pełni twardości mineralnej. Dla czytelnika nieprofesjonalnego wniosek jest taki, że praca mapuje łańcuch zdarzeń od mutacji genu, przez osłabioną adhezję komórek i błędnie okablowane sygnały wzrostu, aż do kruchego szkliwa. Sugeruje też, że precyzyjne dostrojenie tych sygnałów mogłoby pewnego dnia pomóc chronić lub naprawić szkliwo u osób z dziedzicznymi wadami zębów.
Cytowanie: Li, P., Zeng, R., Xue, J. et al. Kdf1 missense mutation caused enamel defects by disrupting cell adhesion and Hippo-YAP signaling in dental epithelium. Int J Oral Sci 18, 43 (2026). https://doi.org/10.1038/s41368-026-00445-4
Słowa kluczowe: rozwój szkliwa, genetyka zębów, adhezja komórek, sygnalizacja Hippo YAP, amelogenesis imperfecta