Clear Sky Science · fr
La mutation faux-sens de Kdf1 provoque des défauts de l’émail en perturbant l’adhésion cellulaire et la voie Hippo‑YAP dans l’épithélium dentaire
Pourquoi une petite protéine dentaire compte
L’émail dentaire est l’armure blanche et brillante qui protège nos dents contre une vie de mastication, de café brûlant et de crème glacée froide. Dans certaines familles, cette armure ne se forme jamais complètement, laissant des dents fragiles, sensibles et sujettes aux caries, une affection appelée amélogenèse imparfaite. Cette étude retrace ce problème jusqu’à une modification subtile d’une seule protéine nommée KDF1 dans les cellules qui construisent l’émail, révélant comment un petit changement génétique peut affaiblir à la fois la coopération cellulaire et les signaux qui guident le développement dentaire.
Du bouclier robuste au revêtement fragile
L’émail est produit par des cellules spécialisées de surface appelées améloblastes, qui sécrètent et durcissent de manière contrôlée une couche riche en protéines sur les dents en croissance. Les chercheurs ont étudié une mutation dérivée d’un patient du gène KDF1 qui avait été liée à des dents manquantes et à un émail de mauvaise qualité. À l’aide de souris génétiquement modifiées pour porter la même mutation, ils ont constaté que les mutants à une copie (hétérozygotes) et à deux copies (homozygotes) développaient un émail plus fin, avec une teneur minérale réduite et un motif interne désorganisé. L’imagerie a montré des couronnes dentaires plus petites, un volume d’émail diminué et une densité d’émail plus faible, en particulier chez les animaux portant deux copies mutantes.
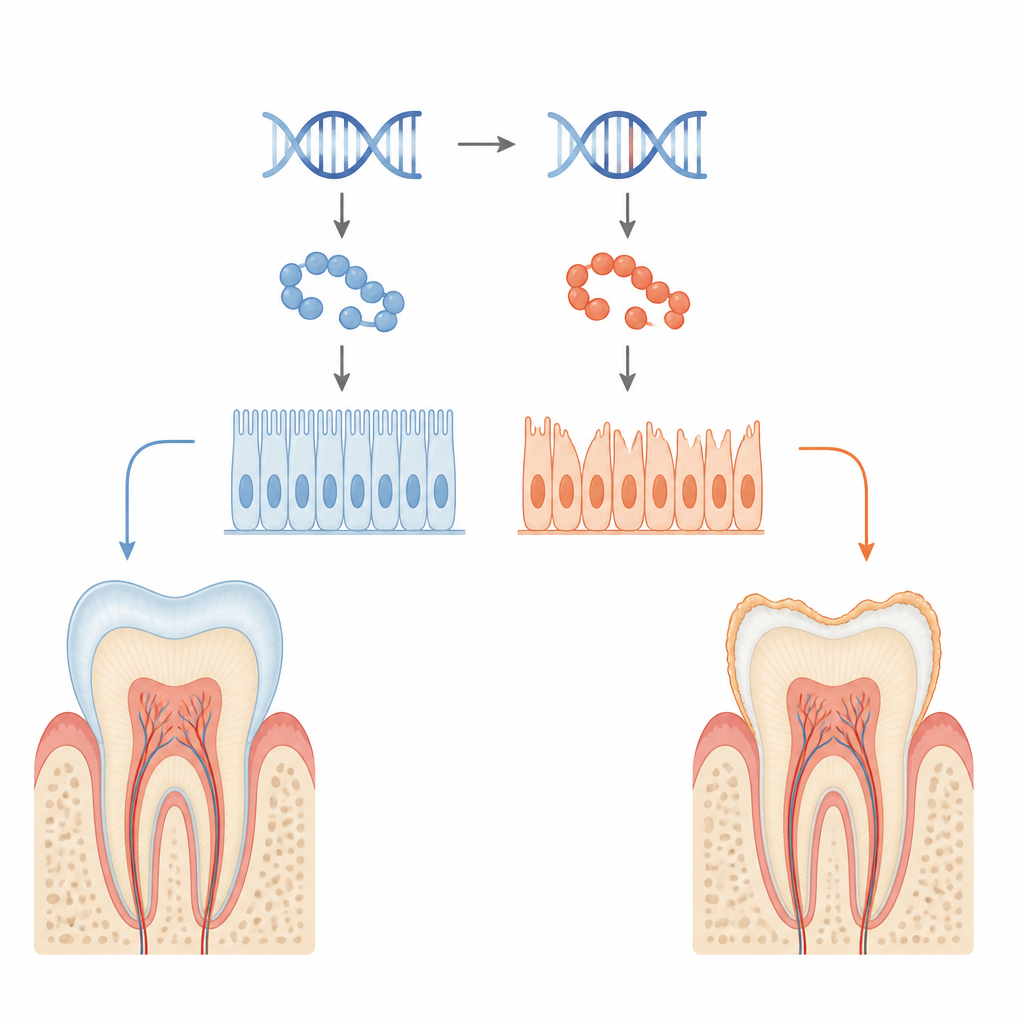
Quand les cellules formatrices d’émail perdent leur prise
En examinant de plus près la surface dentaire, l’équipe a observé que les améloblastes mutants ne s’agrippaient pas fermement les uns aux autres ni à la couche d’émail en formation. La microscopie a révélé des espaces là où les cellules devraient être solidement attachées, et les images ultrastructurales ont montré que les jonctions en ceinture normales entre cellules voisines étaient raccourcies et étirées. Des molécules d’adhésion clés, qui jouent le rôle de rivets biologiques, comme l’E‑cadhérine et l’intégrine β4, étaient nettement réduites chez les animaux mutants. Dans des lignées cellulaires productrices d’émail en culture génétiquement modifiées pour exprimer la KDF1 mutante, les cellules adhéraient moins bien, migraient plus facilement et présentaient une prolifération accrue mais des signes de maturation plus faibles, faisant écho aux défauts observés au niveau tissulaire.
Les feux de signalisation déséquilibrés
L’adhésion cellulaire n’est pas que mécanique ; elle alimente aussi des réseaux de signalisation internes qui indiquent aux cellules quand croître et quand se spécialiser. Le séquençage ARN des améloblastes de souris mutantes a mis en évidence des changements larges dans des gènes liés à la matrice extracellulaire, à l’adhésion et à une voie connue sous le nom de Hippo‑YAP, qui contribue au contrôle de la taille des organes et du destin cellulaire. Dans des dents saines, cette voie maintient la protéine YAP majoritairement inactive dans le cytoplasme, limitant les signaux de croissance. Chez les mutants, YAP était moins phosphorylée, s’accumulait dans le noyau et formait davantage de complexes avec sa partenaire TEAD1, activant des gènes liés à la croissance. Ce schéma correspondait à l’augmentation observée de la division cellulaire dans des régions qui ralentissent normalement à mesure que les cellules de l’émail mûrissent.
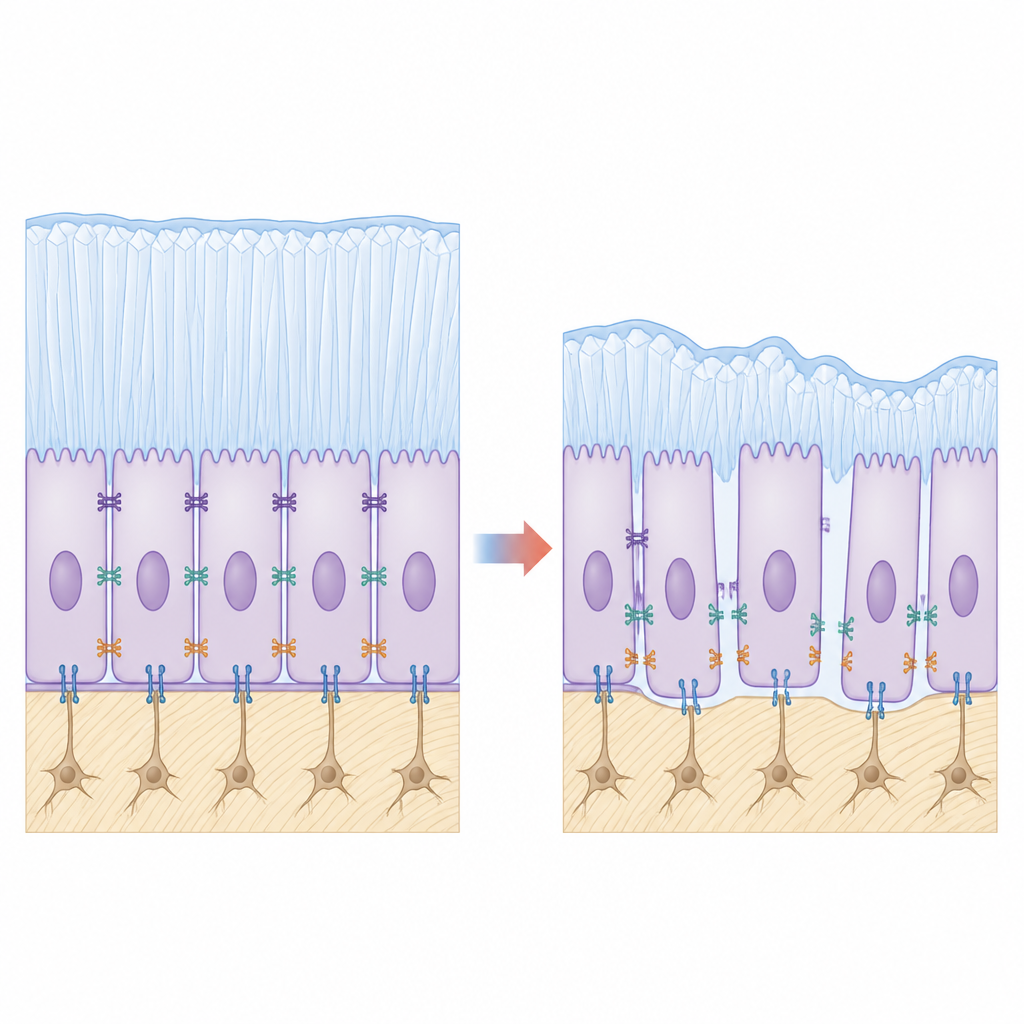
Relier les contacts affaiblis aux signaux défaillants
Pour relier ces éléments, l’équipe a diminué ou augmenté expérimentalement les protéines d’adhésion dans des modèles cellulaires. La réduction de l’E‑cadhérine ou de l’intégrine β4 dans des cellules par ailleurs normales affaiblissait l’activité Hippo‑YAP de façon similaire à la mutation KDF1, tandis que la restauration de ces molécules d’adhésion dans des cellules mutantes ravivait partiellement la phosphorylation de YAP, ramenant le système vers l’équilibre. Cela soutient un modèle dans lequel KDF1 ancré en membrane stabilise les complexes d’adhésion ; quand KDF1 est mutée et mal localisée, ces complexes se désagrègent, les « freins » Hippo‑YAP lâchent, les cellules continuent de se diviser et ne transitionnent pas complètement en spécialistes de la fabrication de l’émail.
Un indice pour une voie thérapeutique future
Les chercheurs ont également testé la vertéporfine, un médicament existant qui interfère avec le partenariat YAP‑TEAD1. Dans des cultures de cellules formatrices d’émail, la vertéporfine a atténué la prolifération excessive et amélioré des marqueurs de production de matrice émail. Chez de jeunes souris mutantes, un traitement précoce par vertéporfine a augmenté le volume d’émail, bien qu’il n’ait pas complètement restauré la dureté minérale. Pour le lecteur non spécialiste, la conclusion est que ce travail cartographie une chaîne allant de la mutation génétique à l’affaiblissement de l’adhésion cellulaire, en passant par des signaux de croissance mal réglés et, enfin, à un émail fragile. Il suggère aussi qu’un ajustement soigneux de ces signaux pourrait un jour aider à protéger ou réparer l’émail chez des personnes porteuses de défauts dentaires héréditaires.
Citation: Li, P., Zeng, R., Xue, J. et al. Kdf1 missense mutation caused enamel defects by disrupting cell adhesion and Hippo-YAP signaling in dental epithelium. Int J Oral Sci 18, 43 (2026). https://doi.org/10.1038/s41368-026-00445-4
Mots-clés: développement de l’émail, génétique dentaire, adhésion cellulaire, signalisation Hippo YAP, amélogenèse imparfaite