Clear Sky Science · en
Kdf1 missense mutation caused enamel defects by disrupting cell adhesion and Hippo-YAP signaling in dental epithelium
Why a small tooth protein matters
Tooth enamel is the glossy white armor that protects our teeth from a lifetime of chewing, hot coffee and cold ice cream. In some families, this armor never fully forms, leaving teeth fragile, sensitive and prone to decay, a condition known as amelogenesis imperfecta. This study traces that problem back to a subtle change in a single protein called KDF1 in the cells that build enamel, revealing how a tiny genetic change can weaken both cell teamwork and the signals that guide tooth development.
From sturdy shield to fragile coating
Enamel is made by specialized surface cells called ameloblasts, which carefully secrete and harden a protein-rich layer on growing teeth. The researchers studied a patient-derived mutation in the KDF1 gene that had been linked to missing teeth and poor enamel. Using mice engineered to carry the same mutation, they found that both one-copy (heterozygous) and two-copy (homozygous) mutants developed thinner enamel with reduced mineral content and a disorganized internal pattern. Imaging showed smaller tooth crowns, less enamel volume and lower enamel density, especially in animals carrying two mutant copies.
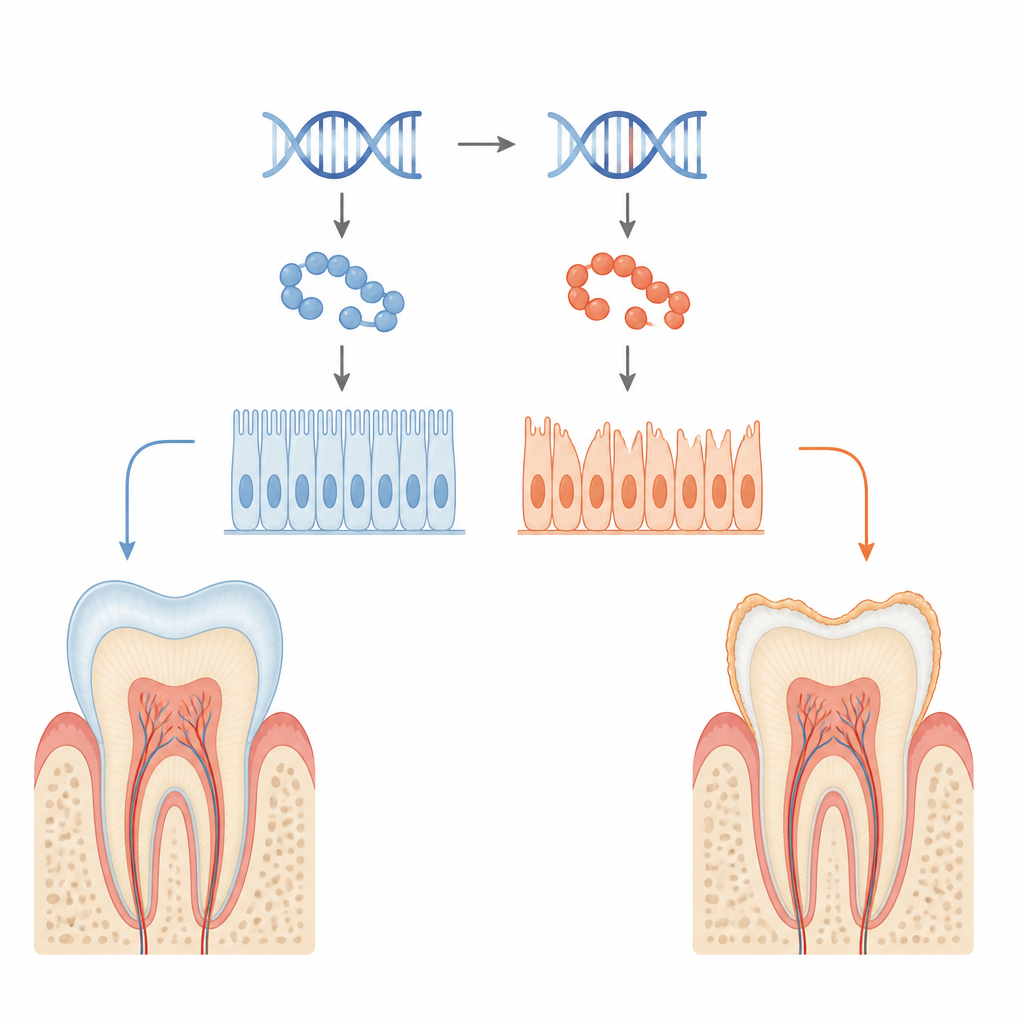
When tooth-building cells lose their grip
Looking more closely at the tooth surface, the team saw that mutant ameloblasts did not cling tightly to each other or to the forming enamel layer. Microscopy revealed gaps where cells should be firmly attached, and ultrastructural images showed that the normal belt-like junctions between neighboring cells were shortened and stretched. Key adhesion molecules that act like biological rivets, such as E-cadherin and integrin β4, were markedly reduced in mutant animals. In cultured enamel-forming cell lines engineered to produce mutant KDF1, cells stuck less well, migrated more readily and showed higher proliferation but poorer signs of maturation, echoing the tissue-level defects.
Signal traffic lights thrown off balance
Cell adhesion is not just mechanical; it also feeds into internal signaling networks that tell cells when to grow and when to specialize. RNA sequencing of ameloblasts from mutant mice pointed to broad changes in genes related to the extracellular matrix, adhesion and a pathway known as Hippo-YAP, which helps control organ size and cell fate. In healthy teeth, this pathway keeps the YAP protein mostly inactive in the cell cytoplasm, limiting growth signals. In the mutants, YAP was less phosphorylated, accumulated in the nucleus and formed more complexes with its partner TEAD1, turning on growth-related genes. This pattern matched the observed surge in cell division in regions that normally slow down as enamel cells mature.
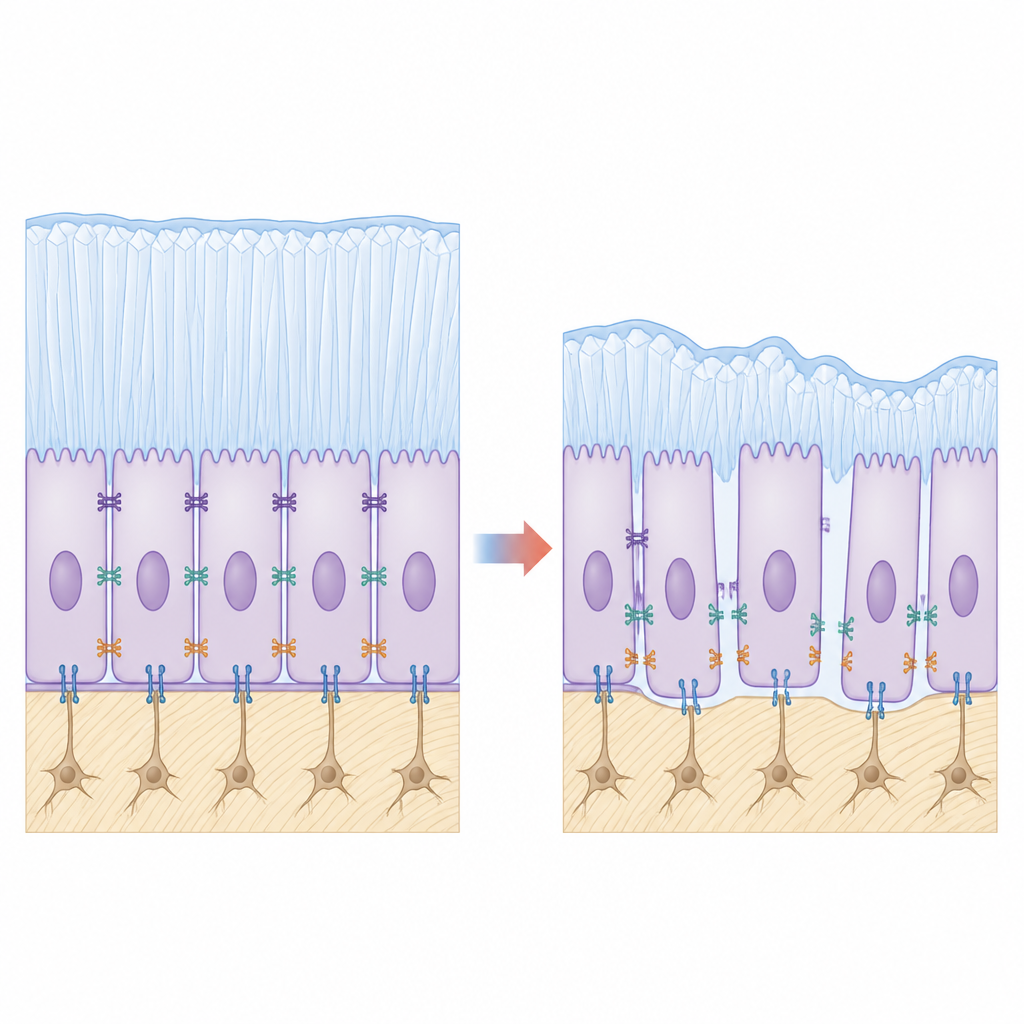
Linking weakened contacts to faulty signals
To connect these dots, the team experimentally lowered or boosted the adhesion proteins in cell models. Reducing E-cadherin or integrin β4 in otherwise normal cells weakened Hippo-YAP activity in a similar way to the KDF1 mutation, while restoring these adhesion molecules in mutant cells partially revived YAP phosphorylation, nudging the system back toward balance. This supports a model in which membrane-bound KDF1 stabilizes adhesion complexes; when KDF1 is mutated and mislocalized, these complexes fall apart, the Hippo-YAP "brakes" slip, cells keep dividing and do not fully transition into enamel-making specialists.
A hint of a future treatment path
The researchers also tested verteporfin, an existing drug that interferes with the YAP-TEAD1 partnership. In enamel-forming cell cultures, verteporfin dampened excessive proliferation and improved markers of enamel matrix production. In young mutant mice, early verteporfin treatment increased enamel volume, although it did not fully restore mineral hardness. For a lay reader, the takeaway is that this work maps a chain from gene mutation, to weakened cell adhesion, to miswired growth signals and, finally, to fragile enamel. It also suggests that carefully tuning these signals might one day help protect or repair enamel in people with inherited tooth defects.
Citation: Li, P., Zeng, R., Xue, J. et al. Kdf1 missense mutation caused enamel defects by disrupting cell adhesion and Hippo-YAP signaling in dental epithelium. Int J Oral Sci 18, 43 (2026). https://doi.org/10.1038/s41368-026-00445-4
Keywords: enamel development, tooth genetics, cell adhesion, Hippo YAP signaling, amelogenesis imperfecta