Clear Sky Science · it
Varianti bialleliche in RNU2-2 causano un’encefalopatia evolutiva ed epilettica sorprendentemente frequente
Perché un gene minuscolo è importante per il cervello dei bambini
I disturbi del neurosviluppo, come il deficit intellettivo e l’epilessia, colpiscono milioni di bambini nel mondo; eppure per circa la metà delle famiglie coinvolte i test genetici non riescono ancora a identificare una causa chiara. Questo studio mostra che alterazioni in un frammento di materiale genetico insolitamente piccolo, un gene di RNA non codificante chiamato RNU2-2, sono alla base di un disturbo cerebrale infantile grave e sorprendentemente comune, caratterizzato da ritardo nello sviluppo precoce e crisi difficili da trattare.
Dai casi irrisolti a un colpevole nascosto
I ricercatori sono partiti da decine di migliaia di persone sottoposte a sequenziamento dell’intero genoma in grandi progetti nazionali e internazionali, molti dei quali bambini con problemi di apprendimento o epilessia non spiegati. Si sono concentrati su una classe speciale di geni che non producono proteine ma generano piccole molecole di RNA che aiutano a assemblare la macchina di splicing della cellula — il sistema che modifica i messaggi di RNA grezzi. Scansionando 1.901 di questi piccoli geni di RNA alla ricerca di varianti rare ereditate da entrambi i genitori, hanno scoperto che un gene, RNU2-2, si distingueva. I bambini con disturbi del neurosviluppo non risolti presentavano coppie rare di varianti in RNU2-2 molto più spesso del previsto rispetto a decine di migliaia di genomi di controllo.
Si profila una nuova sindrome recessiva
Per confermare che non si trattava di un artefatto statistico, il team ha raccolto casi aggiuntivi da banche dati sulle malattie rare nel Regno Unito, in Europa, in Medio Oriente e in Asia. Complessivamente hanno identificato 84 individui affetti provenienti da 67 famiglie con varianti rare “bialleliche” in RNU2-2 — sia la stessa alterazione su entrambe le copie del gene sia due cambiamenti differenti dannosi. Nonostante la raccolta globale, questi bambini mostravano quadri clinici sorprendentemente simili: ritardo dello sviluppo profondo, grave disabilità intellettiva e crisi che spesso esordivano in età infantile. Una larga parte non ha mai imparato a parlare o a camminare autonomamente, e le immagini cerebrali mostravano comunemente perdita di tessuto cerebrale e spazi fluidi ingranditi, coerenti con un’encefalopatia, cioè una disfunzione cerebrale globale.
Come un piccolo RNA compromette la funzione cerebrale
RNU2-2 produce un RNA corto che fa parte dello spliceosoma maggiore, la macchina cellulare che taglia e incolla pezzi di RNA prima che vengano tradotti in proteine. Gli esseri umani possiedono anche una versione di “riserva” molto simile, U2-1. Analizzando l’RNA da campioni di sangue, gli scienziati hanno osservato che i bambini con varianti bialleliche in RNU2-2 avevano quantità marcatamente ridotte dell’RNA U2-2, mentre i livelli di U2-1 rimanevano relativamente stabili. Più importante, il rapporto U2-2/U2-1 era sistematicamente molto più basso negli individui affetti rispetto a qualsiasi controllo, rendendo questo squilibrio un promettente marcatore diagnostico. I dati suggeriscono che molte delle varianti che causano la malattia destabilizzano l’RNA U2-2, riducendone l’abbondanza a un livello che il cervello in sviluppo non può tollerare, anche se U2-1 compensa parzialmente.
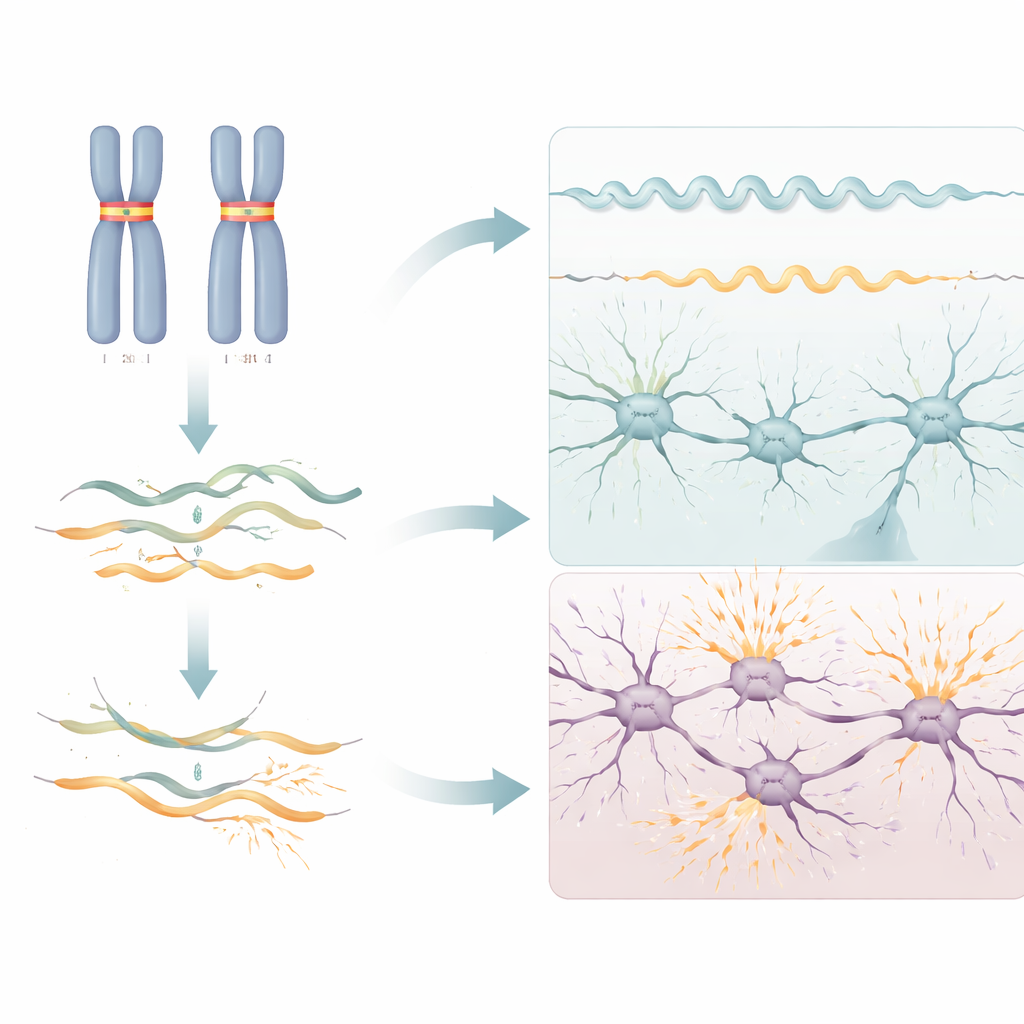
Distinte patologie dallo stesso gene
Era già noto che RNU2-2 può causare una condizione del neurosviluppo di tipo dominante quando solo una copia del gene porta specifiche mutazioni. Il nuovo lavoro dimostra che la sindrome recessiva qui descritta non è semplicemente una forma più lieve o più precoce di quella condizione. Le varianti dannose si distribuiscono su una gamma più ampia di posizioni nell’RNA, le alterazioni dell’RNA sono diverse — nei casi dominanti si osserva un’ampia alterazione dello splicing nel sangue, mentre nei casi recessivi si osserva prevalentemente un deplezione dell’RNA — e le caratteristiche cliniche si sovrappongono solo in parte. Per esempio, le crisi nel primo anno di vita e la rigidità muscolare (spasticità) sono molto più comuni nella forma recessiva, mentre movimenti delle mani distintivi e aspetti del volto sono più tipici della forma dominante.
Quanto è comune questa condizione nascosta?
Quando i ricercatori hanno confrontato RNU2-2 con tutti gli altri geni recessivi noti nel loro ampio gruppo britannico di disturbi del neurosviluppo, la sindrome recessiva da RNU2-2 è emersa come la singola diagnosi recessiva più frequente, superando il gene successivo per diffusione di oltre tre volte. Insieme a condizioni correlate causate da varianti in altri piccoli geni di RNA, questa famiglia di “RNU‑opatie” rappresenta quasi l’1,5% dei casi di neurosviluppo precedentemente irrisolti nel 100,000 Genomes Project — un contributo inaspettatamente consistente da geni che insieme occupano solo poche centinaia di basi di DNA.
Cosa significa per le famiglie e per le cure future
Per le famiglie, la scoperta di questa sindrome recessiva da RNU2-2 implica che una risposta genetica chiara è ora raggiungibile per molti bambini con grave ritardo dello sviluppo ed epilessia. Poiché sono richieste due copie alterate, i genitori sono di solito portatori sani, e test accurati possono guidare le decisioni riproduttive e identificare fratelli a rischio. Per clinici e ricercatori, il lavoro evidenzia l’importanza dei piccoli geni non codificanti che un tempo erano trascurati nei test standard. Indica inoltre una firma molecolare misurabile — il rapporto abbassato U2-2/U2-1 dell’RNA — che può aiutare a confermare le diagnosi e, nel lungo termine, potrebbe orientare gli sforzi per progettare terapie volte a ripristinare il delicato equilibrio di RNA necessario per un sano sviluppo cerebrale.
Citazione: Jackson, A., Blakes, A.J.M., Alhaddad, B. et al. Biallelic variants in RNU2-2 cause a remarkably frequent developmental and epileptic encephalopathy. Nat Genet 58, 798–809 (2026). https://doi.org/10.1038/s41588-026-02551-9
Parole chiave: disturbi del neurosviluppo, encefalopatia epilettica, RNA non codificante, spliceosoma, sequenziamento del genoma