Clear Sky Science · fr
Des variants bialléliques dans RNU2-2 causent une encéphalopathie développementale et épileptique remarquablement fréquente
Pourquoi un gène minuscule compte pour le cerveau des enfants
Les troubles du neurodéveloppement tels que la déficience intellectuelle et l’épilepsie touchent des millions d’enfants dans le monde ; pourtant, pour environ la moitié des familles concernées, les tests génétiques ne parviennent toujours pas à identifier une cause claire. Cette étude révèle que des altérations d’un fragment de matériel génétique exceptionnellement petit, un gène d’ARN non codant nommé RNU2-2, sous-tendent un trouble cérébral infantile sévère et étonnamment fréquent, caractérisé par un retard du développement précoce et des crises difficiles à traiter.
Des cas non résolus à un coupable caché
Les chercheurs ont commencé par des dizaines de milliers de personnes ayant bénéficié d’un séquençage du génome complet dans le cadre de grands projets nationaux et internationaux, beaucoup d’entre elles étant des enfants présentant des troubles d’apprentissage ou une épilepsie d’étiologie inconnue. Ils se sont focalisés sur une classe particulière de gènes qui ne codent pas de protéines mais produisent de petits ARN aidant à assembler la machinerie d’épissage de la cellule — le système qui édite les messages ARN bruts. En examinant 1 901 de ces petits gènes d’ARN à la recherche de variants rares hérités des deux parents, ils ont identifié un gène, RNU2-2, qui se démarquait. Les enfants présentant des troubles du neurodéveloppement non résolus portaient des paires rares de variants RNU2-2 beaucoup plus souvent qu’attendu par comparaison avec des dizaines de milliers de génomes témoins.
Un nouveau syndrome récessif prend forme
Pour vérifier qu’il ne s’agissait pas d’un artefact statistique, l’équipe a rassemblé des cas supplémentaires issus de bases de données de maladies rares au Royaume-Uni, en Europe, au Moyen-Orient et en Asie. Au total, ils ont identifié 84 personnes affectées issues de 67 familles présentant des variants rares « bialléliques » de RNU2-2 — soit le même changement sur les deux copies du gène, soit deux changements délétères différents. Bien que collectés dans le monde entier, ces enfants présentaient des tableaux cliniques remarquablement similaires : retard global du développement, déficience intellectuelle sévère et, dans de nombreux cas, crises débutant dès l’enfance. Une grande proportion n’a jamais appris à parler ni à marcher de manière autonome, et les imageries cérébrales montraient fréquemment une perte de tissu cérébral et une dilatation des espaces liquidiens, cohérentes avec une encéphalopathie, c’est‑à‑dire un dysfonctionnement cérébral global.
Comment un petit ARN perturbe la fonction cérébrale
RNU2-2 produit un court ARN qui fait partie du spliceosome majeur, la machine cellulaire qui recoupe et assemble des fragments d’ARN avant qu’ils ne soient traduits en protéines. L’humain possède également une copie « de secours » très similaire, U2-1. En analysant l’ARN provenant d’échantillons sanguins, les chercheurs ont constaté que les enfants porteurs de variants bialléliques de RNU2-2 présentaient des quantités nettement réduites de l’ARN U2-2, tandis que les niveaux de U2-1 restaient relativement stables. Plus important encore, le rapport U2-2/U2-1 était systématiquement beaucoup plus bas chez les sujets affectés que chez les témoins, faisant de ce déséquilibre un marqueur diagnostique prometteur. Les données suggèrent que bon nombre des variants pathogènes déstabilisent l’ARN U2-2, réduisant son abondance à un niveau que le cerveau en développement ne peut tolérer, même si U2-1 compense partiellement.
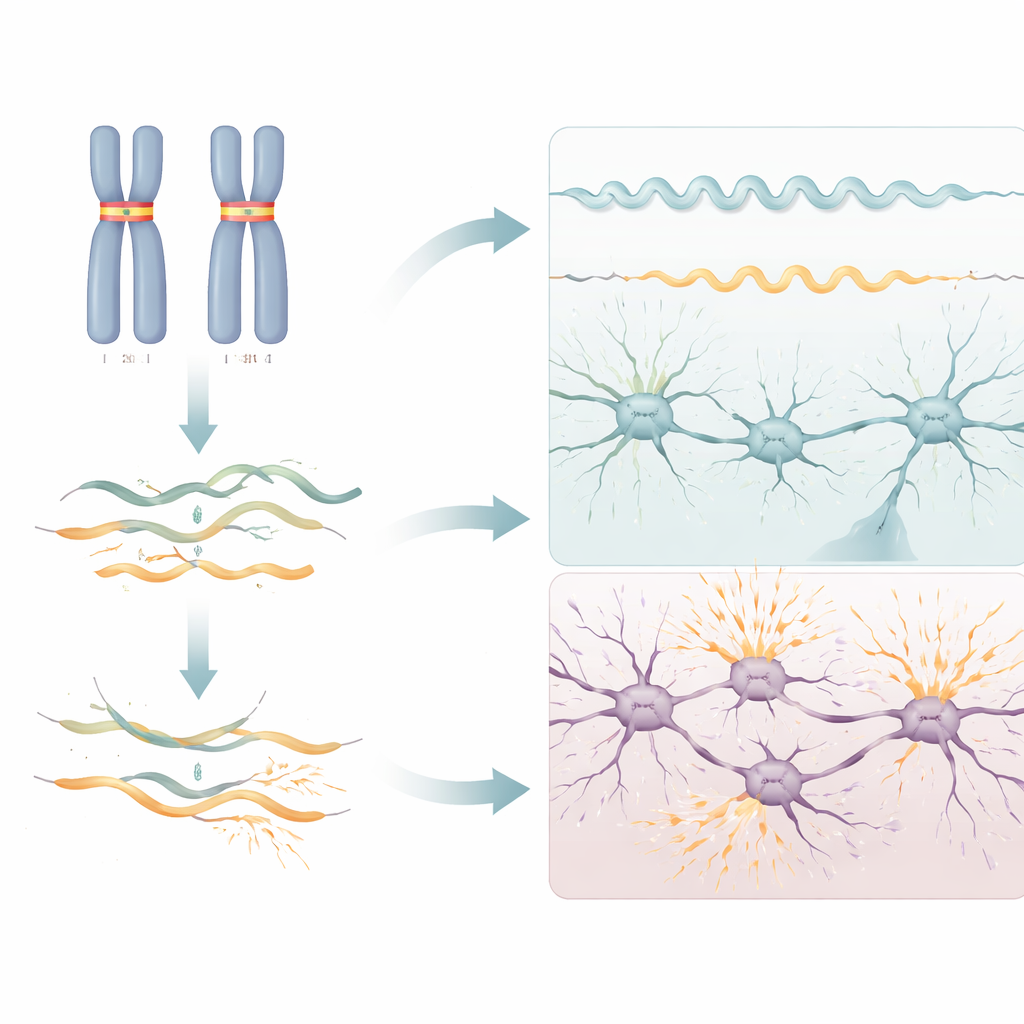
Des troubles distincts issus du même gène
On savait déjà que RNU2-2 peut provoquer une autre maladie neurodéveloppementale de type dominant lorsque seule une copie du gène porte certaines mutations spécifiques. Les travaux récents montrent que le syndrome récessif mis au jour ici n’est pas simplement une forme atténuée ou plus précoce de cette maladie dominante. Les variants délétères se situent sur une gamme de positions plus large dans l’ARN, les altérations de l’ARN diffèrent — les cas dominants présentent une perturbation généralisée de l’épissage dans le sang, tandis que les cas récessifs montrent principalement une déplétion de l’ARN — et les caractéristiques cliniques ne se recoupent que partiellement. Par exemple, les crises dans la première année de vie et la raideur musculaire (spasticité) sont beaucoup plus fréquentes dans la forme récessive, alors que des mouvements manuels distinctifs et une apparence faciale particulière sont plus typiques de la forme dominante.
Quelle est la fréquence de cette affection cachée ?
Lorsque les chercheurs ont comparé RNU2-2 à tous les autres gènes récessifs connus dans leur large cohorte britannique de troubles du neurodéveloppement, le syndrome récessif lié à RNU2-2 est apparu comme le diagnostic récessif le plus fréquent, dépassant le gène suivant le plus courant de plus de trois fois. Avec des affections apparentées causées par des variants dans d’autres petits gènes d’ARN, cette famille d’« RNU‑opathies » représente près de 1,5 % des cas de neurodéveloppement auparavant non résolus dans le projet 100 000 Genomes — une contribution étonnamment élevée de gènes qui occupent ensemble seulement quelques centaines de lettres d’ADN.
Ce que cela signifie pour les familles et les soins futurs
Pour les familles, la découverte de ce syndrome récessif RNU2-2 signifie qu’une réponse génétique claire est désormais à la portée de nombreux enfants souffrant de retard sévère du développement et d’épilepsie. Parce que deux copies altérées sont requises, les parents sont généralement des porteurs sains, et un dépistage précis peut orienter les décisions reproductives et identifier des frères et sœurs à risque. Pour les cliniciens et les chercheurs, ce travail souligne l’importance des petits gènes non codants autrefois négligés dans les tests standards. Il met aussi en évidence une signature moléculaire mesurable — la baisse du ratio ARN U2-2/U2-1 — qui peut aider à confirmer les diagnostics et, à plus long terme, pourrait informer des efforts visant à concevoir des thérapies rétablissant l’équilibre délicat des ARN nécessaire au développement cérébral sain.
Citation: Jackson, A., Blakes, A.J.M., Alhaddad, B. et al. Biallelic variants in RNU2-2 cause a remarkably frequent developmental and epileptic encephalopathy. Nat Genet 58, 798–809 (2026). https://doi.org/10.1038/s41588-026-02551-9
Mots-clés: troubles du neurodéveloppement, encéphalopathie épileptique, ARN non codant, spliceosome, séquençage du génome