Clear Sky Science · fr
Estimation efficace en données des fonctions de partition dans les matériaux désordonnés par IA
Pourquoi de minuscules défauts dans des matériaux désordonnés comptent
Beaucoup des matériaux les plus prometteurs d’aujourd’hui — des carburants nucléaires aux alliages haute performance — ne sont pas des cristaux parfaitement ordonnés mais des « patchworks » chimiques où différents atomes sont fortement mélangés. Dans ces structures désordonnées, de minuscules atomes manquants et autres défauts contrôlent la manière dont la chaleur, l’électricité et même les dommages par radiation se propagent dans le matériau. L’article présente PULSE, un nouvel outil d’intelligence artificielle capable de prédire le comportement de tels défauts dans des matériaux complexes et désordonnés bien plus efficacement que les simulations informatiques traditionnelles.
Le problème de compter l’à‑peu‑près incalculable
Dans des matériaux multicomposants comme les oxydes mixtes uranium‑plutonium ou les alliages à haute entropie, les atomes peuvent occuper des sites du réseau d’un très grand nombre de manières. Chaque arrangement distinct, ou configuration, peut modifier légèrement la facilité de formation et de déplacement des défauts. Pour prédire les propriétés réelles, les chercheurs doivent en principe considérer toutes ces possibilités, ce que capture mathématiquement une quantité appelée fonction de partition. Les approches classiques approximent soit le désordre par une poignée de structures modèles soigneusement conçues, soit échantillonnent de nombreuses configurations par des simulations de Monte‑Carlo. La première stratégie peut passer à côté d’arrangements importants ; la seconde est souvent tellement coûteuse en calcul qu’elle devient impraticable pour des systèmes réalistes.
Un échantillonneur IA qui s’auto‑entraîne
PULSE (Partition function Unsupervised Learning Sampling and Evaluation) relève ce défi en apprenant à explorer l’espace des configurations de manière autonome. Il repose sur un autoencodeur variationnel inverse, un type de réseau neuronal qui part d’entrées aléatoires simples et les transforme en configurations atomiques réalistes. Fondamentalement, ce modèle n’a pas besoin d’une base de données d’exemples précompilée. Il génère des arrangements candidats, les envoie à un calculateur atomistique pour obtenir leurs énergies, puis ajuste ses paramètres internes pour rendre les échantillons utiles plus probables. Cette boucle fermée permet à l’IA de concentrer son attention sur les configurations qui comptent le plus pour la fonction de partition, en particulier celles qui dominent le comportement à une température donnée.
Tester PULSE sur du carburant nucléaire
Pour démontrer ses capacités, les auteurs appliquent PULSE à l’oxyde uranium‑plutonium, un matériau clé du combustible nucléaire. Ils se concentrent sur les défauts de Schottky — groupes d’atomes manquants qui influencent fortement la réponse du combustible à haute température et aux radiations. D’abord, ils testent la méthode sur des environnements locaux relativement petits où la fonction de partition peut encore être calculée exactement par force brute. Les estimations de PULSE pour cette quantité, et pour les concentrations de défauts qui en résultent, correspondent étroitement aux résultats exacts tout en nécessitant un nombre d’évaluations énergétiques inférieur de plusieurs ordres de grandeur à celui d’un balayage de la base complète. Comparé aux « structures quasi‑aléatoires spéciales » largement utilisées, PULSE obtient une précision similaire ou supérieure, mais avec une mesure de convergence intégrée claire et un coût informatique bien moindre à haute température.
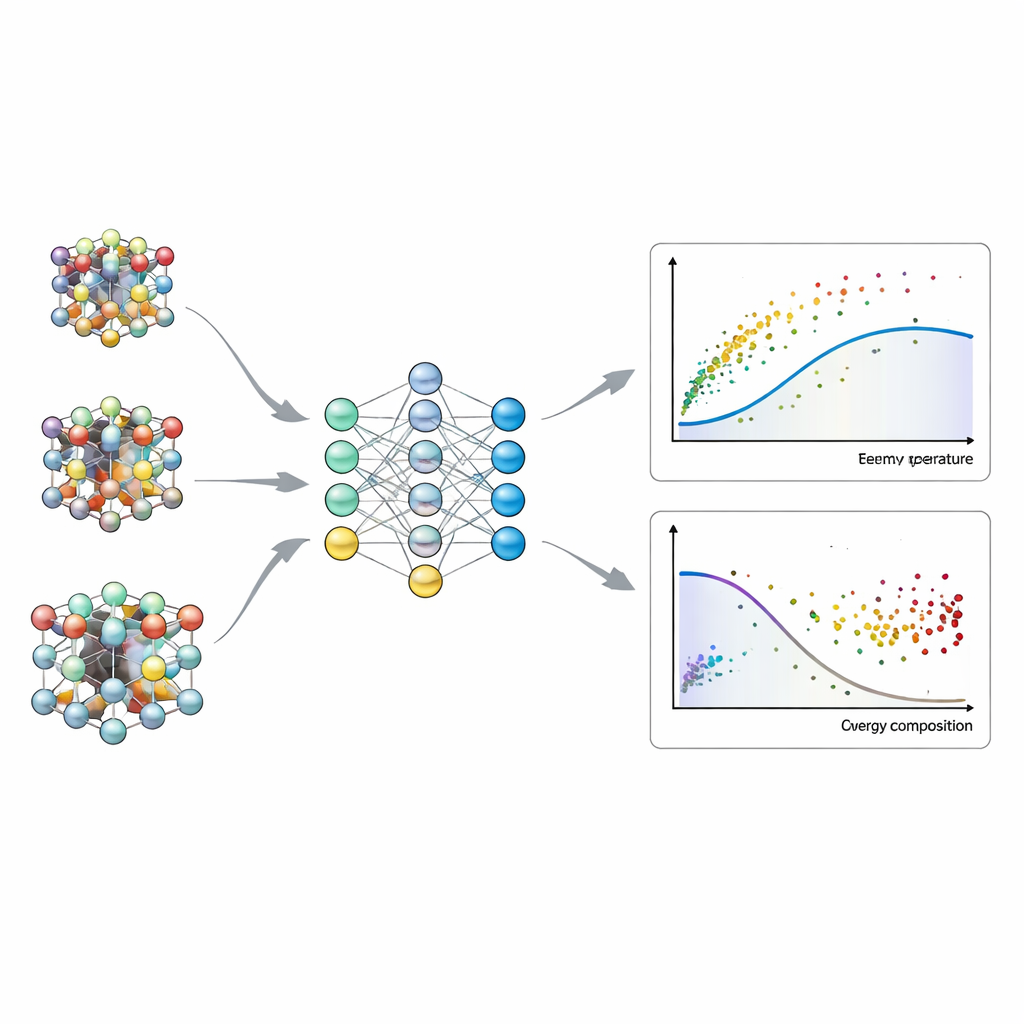
Ce que l’IA révèle sur l’environnement local
Parce que PULSE génère explicitement et évalue des voisinages atomiques autour d’un défaut, il peut être utilisé pour sonder l’étendue de l’influence d’un défaut. En randomisant systématiquement des couches d’atomes de plus en plus éloignées du défaut, les auteurs montrent que, dans cet oxyde combustible, seules quelques couches voisines affectent significativement l’énergétique des défauts, et que la distance pertinente diminue lorsque la température augmente. La méthode montre aussi comment les concentrations de défauts évoluent avec la teneur en plutonium : à mesure que le matériau est enrichi en plutonium, la formation de défauts devient généralement plus facile, conduisant à des densités de défaut plus élevées — en particulier à basses températures où les différences d’énergie sont plus déterminantes.
Pourquoi cela compte pour la conception des matériaux à venir
Pour un non‑spécialiste, le message principal est que PULSE offre un moyen rapide et flexible pour que l’IA « compte » et priorise les arrangements atomiques les plus importants à l’intérieur de matériaux très désordonnés. Plutôt que de simuler exhaustivement chaque configuration possible, la méthode apprend en temps réel quels motifs locaux contrôlent la formation de défauts, puis utilise cette connaissance pour estimer les concentrations de défauts et les propriétés associées. Bien que démontrée sur du carburant nucléaire, la même approche peut s’appliquer à une large gamme de matériaux complexes, tels que les alliages à haute entropie ou les oxydes mixtes pour le stockage d’énergie et la catalyse. De cette manière, PULSE pourrait aider les chercheurs à explorer rapidement comment le désordre microscopique façonne les performances macroscopiques, guidant la conception de matériaux plus sûrs et plus efficaces.
Citation: Karcz, M.J., Messina, L., Kawasaki, E. et al. AI-driven data-efficient estimation of partition functions in disordered materials. Sci Rep 16, 14568 (2026). https://doi.org/10.1038/s41598-026-37953-6
Mots-clés: matériaux désordonnés, apprentissage automatique, carburant nucléaire, défauts ponctuels, modèles génératifs