Clear Sky Science · fr
Aperçus structurels et criblage prédictif du transport ionique dans des alliages riches en lithium via des potentiels par neuroévolution
Pourquoi cela compte pour de meilleures batteries
Les batteries lithium-ion alimentent nos téléphones, nos voitures et une part croissante du réseau électrique. Pourtant, un goulot d'étranglement subsiste : la vitesse à laquelle les ions lithium peuvent se déplacer à l'intérieur des électrodes. Cet article s'attaque à ce problème pour une classe prometteuse de matériaux d'électrode négative — des alliages riches en lithium à base d'indium et d'étain — en utilisant des méthodes avancées d'apprentissage automatique pour observer le mouvement des ions à travers ces labyrinthes atomiques complexes. Les connaissances acquises pourraient aider les ingénieurs à concevoir des batteries qui se chargent plus vite, durent plus longtemps et fonctionnent de façon plus sûre.
Trouver les trajectoires les plus rapides dans les labyrinthes atomiques
Dans les anodes en alliage, le lithium ne se contente pas de se glisser entre les couches ; il forme en réalité de nouveaux composés avec des métaux comme l'indium et l'étain. Ces alliages peuvent atténuer certains problèmes de sécurité du lithium métallique pur, mais introduisent un nouveau casse-tête : les structures atomiques sont complexes, et le lithium peut se déplacer de multiples manières concurrentes. Les auteurs montrent que trois ingrédients contrôlent en grande partie la vitesse de déplacement du lithium. Le premier est le type de « porteur » : une lacune de lithium (vacance), un lithium supplémentaire inséré entre les atomes (interstitiel), ou une lacune sur le site métallique. Le deuxième est la qualité de la connexion des routes à faible résistance à travers le cristal. Le troisième est l'environnement immédiat autour de chaque ion en mouvement, incluant la distance du saut et la manière dont sa charge électrique se modifie en chemin.
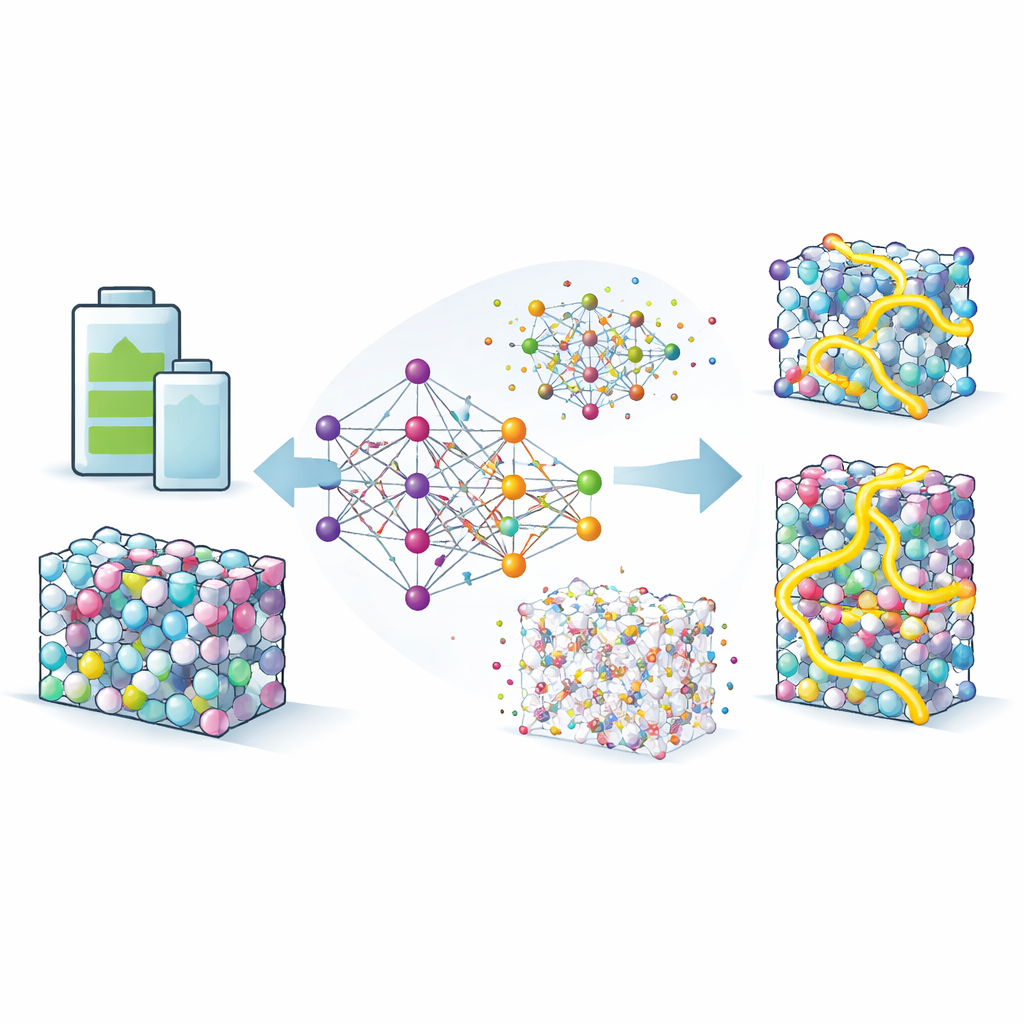
Apprendre un substitut numérique à la physique quantique
Pour explorer ces effets en détail, l'équipe construit des potentiels par neuroevolution, un type de modèle d'apprentissage automatique entraîné à imiter la précision des calculs de mécanique quantique à une fraction du coût. Ils alimentent ce modèle avec des milliers de configurations atomiques tirées de bases de données et de recherches informatiques étendues, puis le raffinant jusqu'à ce qu'il reproduise fidèlement énergies et forces. Avec ce substitut, ils peuvent lancer des simulations longues et à grande échelle dans lesquelles les atomes de lithium se déplacent à travers les alliages Li–In et Li–Sn à des températures réalistes. Le modèle non seulement reproduit les taux de diffusion expérimentaux dans un composé de référence, mais il restitue aussi correctement des propriétés structurelles subtiles, ce qui donne confiance quant à sa capacité à explorer des phases inconnues.
Nouvelles structures d'alliage et leur « respiration »
Armés de ce substitut rapide, les chercheurs effectuent une recherche systématique de composés stables et quasi-stables dans les systèmes lithium–indium et lithium–étain. Ils retrouvent la plupart des phases connues et prédisent plusieurs nouvelles qui paraissent viables sur les plans énergétique et dynamique, suggérant qu'elles pourraient être synthétisées en laboratoire. À mesure que l'on ajoute du lithium, le réseau d'atomes d'indium ou d'étain évolue de maillages tridimensionnels étendus vers des feuillets, des chaînes, puis des atomes isolés, tandis que le lithium cède des électrons et adopte un rôle majoritairement ionique. Les simulations suivent également le gonflement de ces alliages lors de l'absorption de lithium — d'environ deux fois et demie en volume à pleine charge — en accord avec les observations expérimentales et crucial pour évaluer la durabilité mécanique dans des batteries réelles.
Comment le lithium se déplace réellement dans ces alliages
En suivant des atomes individuels en mouvement, l'étude révèle quand et comment différents porteurs dominent. À faible teneur en lithium, des atomes de lithium isolés favorisent l'apparition d'interstitiels supplémentaires en « éjectant » des voisins via des mouvements à faible énergie. À des teneurs plus élevées, des réseaux continus de lithium se forment, et les lacunes de lithium deviennent les principaux porteurs, diffusant le long de canaux connectés. La vitesse globale n'est cependant pas déterminée uniquement par la barrière la plus basse isolée, mais par la présence de nombreux sauts à faible barrière qui se relient en autoroutes de long parcours. Dans certaines structures, un réseau de trajectoires doucement inclinées permet au lithium de traverser le cristal aisément ; dans d'autres, des segments à faible résistance sont enfermés dans des impasses et le mouvement ralentit considérablement. Les alliages à base d'étain reproduisent en grande partie ce comportement, avec des différences subtiles liées à des liaisons légèrement plus fortes.
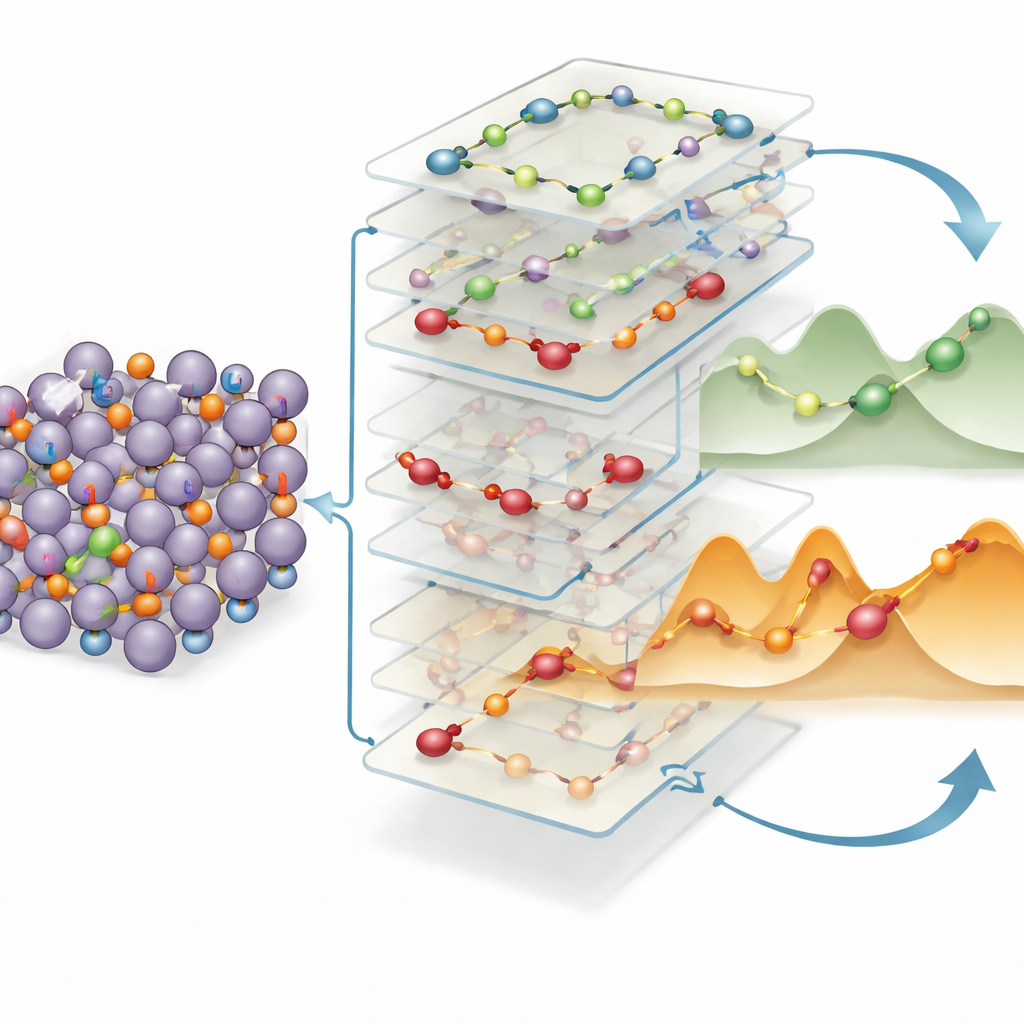
Règles simples pour repérer les autoroutes ioniques rapides
Pour convertir ces connaissances en règles de conception pratiques, les auteurs entraînent un autre modèle d'apprentissage automatique pour relier des descripteurs structurels locaux — distance de saut, amplitude du déplacement de charge, géométrie de liaison locale, et plus — au coût énergétique de chaque mouvement. Ils trouvent que deux facteurs dominent : la distance que doit parcourir le lithium et l'ampleur de la perturbation de sa distribution de charge entre le départ et l'arrivée. Des sauts plus longs et des réarrangements de charge plus importants signifient de manière fiable des barrières plus élevées. De façon frappante, les mêmes motifs favorables — sauts courts le long de chemins bien connectés avec de faibles changements de charge — réapparaissent dans une large gamme d'alliages riches en lithium au-delà de l'indium et de l'étain, y compris des composés contenant du silicium et du germanium. Cette « héritabilité » structurelle suggère que les ingénieurs peuvent cribler des bases de données de matériaux à la recherche de ces motifs pour identifier rapidement de nouveaux anodes en alliage offrant un transport du lithium intrinsèquement rapide.
Ce que cela signifie pour les batteries du futur
En termes simples, ce travail montre que les meilleurs anodes en alliage sont ceux dont l'armature atomique offre au lithium un réseau de pierres d'étape courtes et progressivement graduées plutôt qu'un paysage de mares isolées et de collines abruptes. En combinant des substituts d'apprentissage automatique fidèles à la mécanique quantique avec des règles structurelles claires, l'étude trace une feuille de route pour scanner informatiquement de vastes familles d'alliages riches en lithium avant de les synthétiser en laboratoire. Cela pourrait accélérer la découverte d'électrodes de batterie qui se chargent rapidement, stockent davantage d'énergie et fonctionnent de manière fiable pendant des années d'utilisation.
Citation: Jin, D., Ding, S., Qiu, H. et al. Structural insights and predictive screening of ion transport in Li-rich alloys via neuroevolution potentials. npj Comput Mater 12, 132 (2026). https://doi.org/10.1038/s41524-026-02012-1
Mots-clés: diffusion des ions lithium, anodes en alliage, potentiels d'apprentissage automatique, alliages Li-In et Li-Sn, voies de transport ionique