Clear Sky Science · es
Perspectivas estructurales y cribado predictivo del transporte iónico en aleaciones ricas en Li mediante potenciales por neuroevolución
Por qué importa esto para baterías mejores
Las baterías de ion-litio alimentan nuestros teléfonos, coches y una porción creciente de la red eléctrica. Aun así, persiste un cuello de botella: la rapidez con la que los iones de litio pueden moverse dentro de los electrodos. Este artículo aborda ese problema para una clase prometedora de materiales de cátodo negativo —aleaciones ricas en litio basadas en indio y estaño— usando aprendizaje automático avanzado para observar cómo los iones atraviesan estos complejos laberintos atómicos. Los conocimientos obtenidos podrían ayudar a los ingenieros a diseñar baterías que se carguen más rápido, duren más y funcionen con mayor seguridad.
Encontrar las rutas más rápidas a través de laberintos atómicos
En los ánodos de aleación, el litio no se limita a colarse entre capas; en realidad forma nuevos compuestos con metales como el indio y el estaño. Estas aleaciones pueden mitigar algunos problemas de seguridad del litio metálico puro, pero plantean un nuevo rompecabezas: las estructuras atómicas son intrincadas y el litio puede desplazarse de muchas maneras competitivas. Los autores muestran que tres ingredientes controlan en gran medida la velocidad del litio. Primero, el tipo de “portador” que se mueve: una ausencia de litio (vacante), un litio extra situado entre átomos (intersticial) o la ausencia de un átomo metálico. Segundo, la calidad de la conexión de rutas de baja resistencia a través del cristal. Tercero, el entorno inmediato alrededor de cada ion en movimiento, incluyendo la distancia del salto y cómo varía su carga eléctrica durante el trayecto.
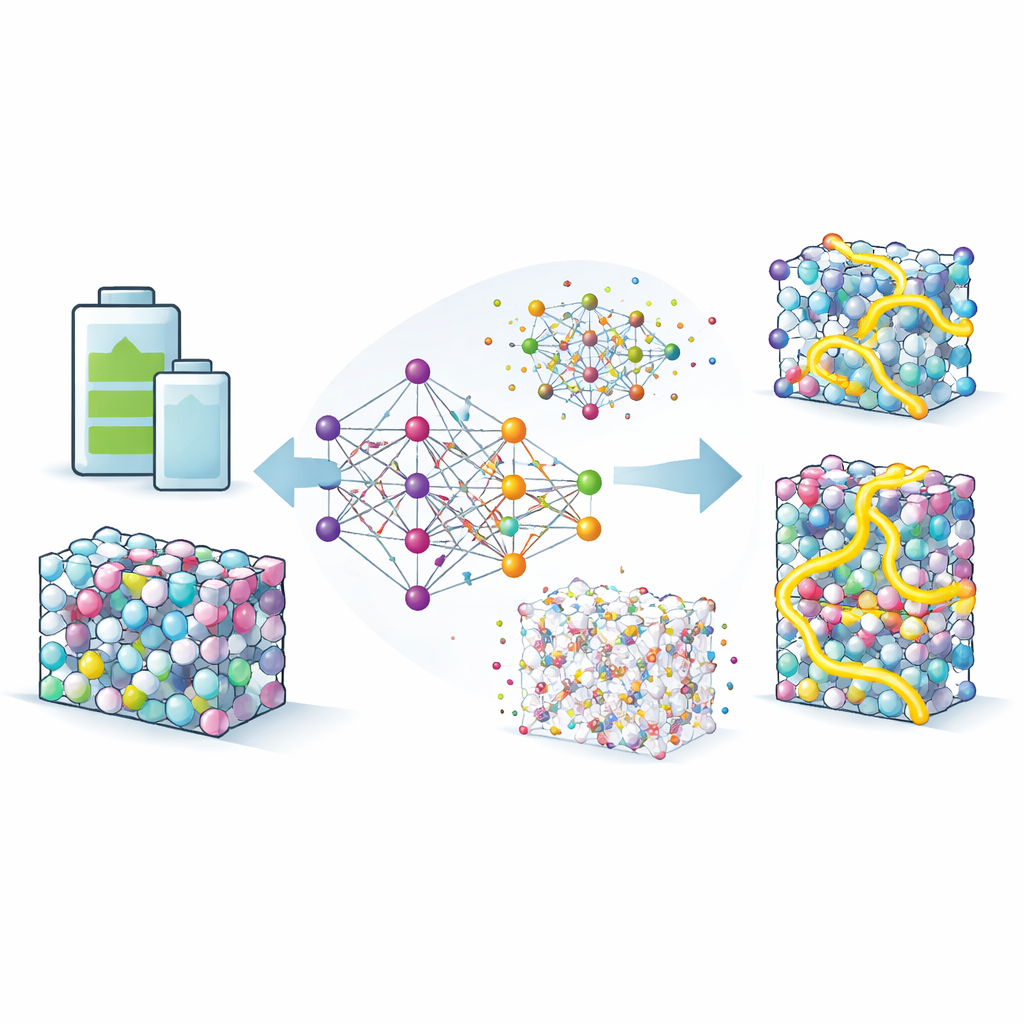
Enseñar a un sustituto digital de la física cuántica
Para explorar estos efectos en detalle, el equipo construye los denominados potenciales por neuroevolución, un tipo de modelo de aprendizaje automático entrenado para imitar la precisión de los cálculos de mecánica cuántica a una fracción del coste. Alimentan este modelo con miles de disposiciones atómicas extraídas de bases de datos y de extensas búsquedas computacionales, y lo refinan hasta que reproduce energías y fuerzas con alta fidelidad. Con este sustituto, pueden ejecutar simulaciones largas y a gran escala en las que los átomos de litio vagan por aleaciones Li–In y Li–Sn a temperaturas realistas. El modelo no solo coincide con experimentos sobre tasas de difusión en un compuesto de referencia, sino que también reproduce correctamente propiedades estructurales sutiles, lo que da confianza para explorar fases poco conocidas.
Nuevas estructuras de aleación y cómo se ‘respiran’
Con el sustituto rápido, los investigadores buscan sistemáticamente compuestos estables y casi estables en los sistemas litio–indio y litio–estaño. Recuperan la mayoría de las fases conocidas previamente y predicen varias nuevas que parecen viables tanto energéticamente como dinámicamente, lo que sugiere que podrían sintetizarse en el laboratorio. A medida que se agrega más litio, la red de átomos de indio o estaño evoluciona desde esqueletos tridimensionales extendidos hacia láminas, cadenas y finalmente átomos aislados, mientras el litio dona electrones y adopta un papel mayoritariamente iónico. Las simulaciones también siguen cuánto se hinchan estas aleaciones al absorber litio —alrededor de dos veces y media en volumen en carga completa—, consistente con observaciones experimentales y crucial para evaluar la durabilidad mecánica en baterías reales.
Cómo se mueve realmente el litio dentro de estas aleaciones
Siguiendo átomos individuales en movimiento, el estudio revela cuándo y cómo dominan diferentes portadores. A bajos contenidos de litio, los átomos de litio aislados favorecen que intersticiales adicionales salten al “desplazar” vecinos mediante movimientos de baja energía. A niveles más altos de litio, se forman redes continuas de litio y las vacantes —sitios de litio ausentes— se convierten en los principales portadores, difundiendo a lo largo de canales conectados. La velocidad global, sin embargo, no la determina solo la barrera individual más baja, sino si muchos saltos de baja barrera se conectan en autopistas de largo alcance. En algunas estructuras, una trama de rutas de pendiente suave permite que el litio atraviese el cristal con facilidad; en otras, los tramos de baja resistencia quedan atrapados en callejones sin salida y el movimiento se ralentiza drásticamente. Las aleaciones a base de estaño reproducen en gran medida este comportamiento, con diferencias sutiles debidas a un enlace algo más fuerte.
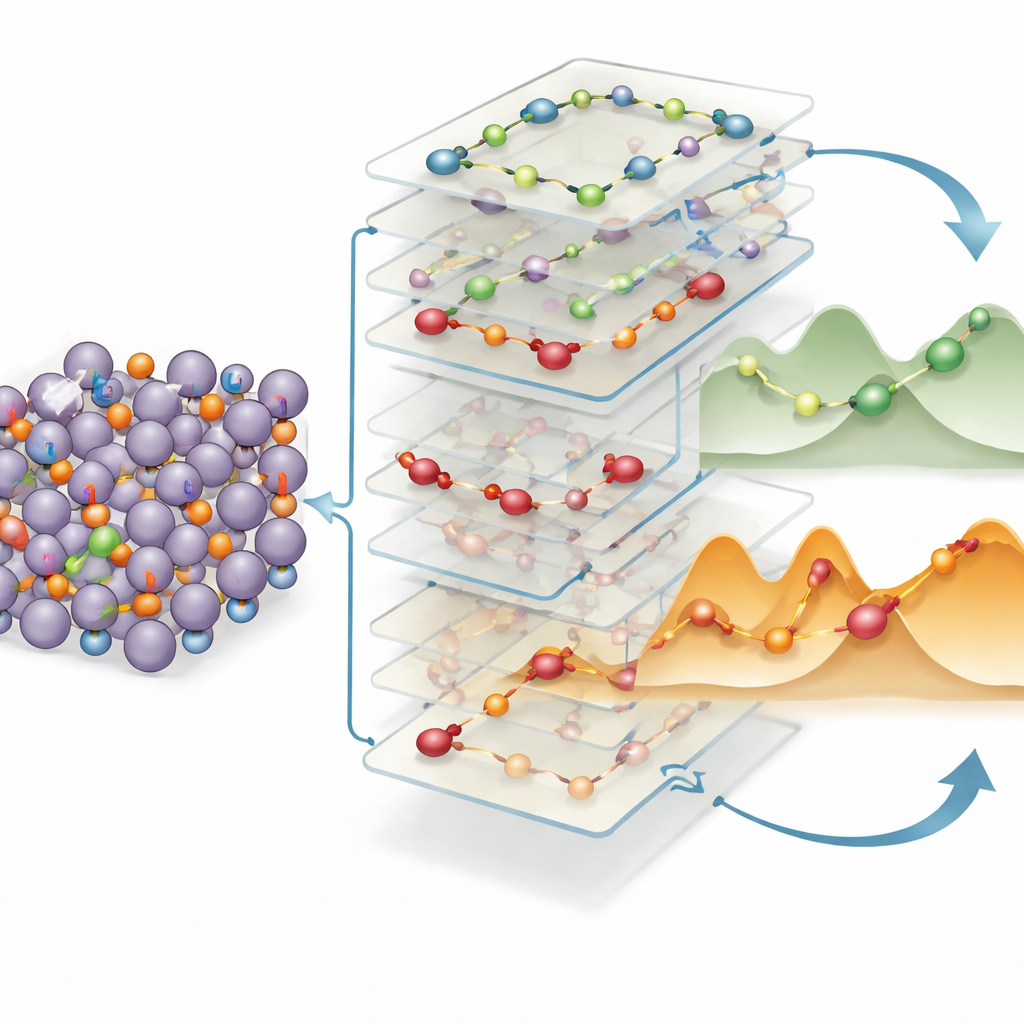
Reglas simples para detectar autopistas iónicas rápidas
Para convertir estos conocimientos en reglas de diseño prácticas, los autores entrenan otro modelo de aprendizaje automático que relaciona descriptores estructurales locales —distancia de salto, cuánto cambia la carga, geometría de enlace local y más— con el coste energético de cada movimiento. Encuentran que dos factores dominan: la distancia que debe saltar el litio y cuánto se altera su distribución de carga entre el inicio y el final. Saltos más largos y grandes redistribuciones de carga implican barreras más altas de forma fiable. Sorprendentemente, los mismos patrones favorables —saltos cortos a lo largo de rutas bien conectadas con cambios de carga suaves— vuelven a aparecer en una amplia gama de aleaciones ricas en litio más allá del indio y el estaño, incluyendo compuestos con silicio y germanio. Esta “herencia” estructural sugiere que los ingenieros pueden cribar bases de datos de materiales buscando estos motivos para identificar rápidamente nuevas aleaciones anódicas con transporte de litio intrínsecamente rápido.
Qué significa esto para las baterías del futuro
En términos cotidianos, este trabajo muestra que los mejores ánodos de aleación son aquellos cuyo entramado atómico ofrece al litio una red de peldaños cortos y suavemente escalonados en lugar de un paisaje de charcos aislados y colinas empinadas. Al combinar sustitutos de mecánica cuántica precisos basados en aprendizaje automático con reglas estructurales claras, el estudio traza una hoja de ruta para escanear por ordenador familias enormes de aleaciones ricas en litio antes de sintetizarlas en el laboratorio. Eso podría acelerar el descubrimiento de electrodos de baterías que se carguen rápido, almacenen más energía y ciclen de forma fiable durante años de uso.
Cita: Jin, D., Ding, S., Qiu, H. et al. Structural insights and predictive screening of ion transport in Li-rich alloys via neuroevolution potentials. npj Comput Mater 12, 132 (2026). https://doi.org/10.1038/s41524-026-02012-1
Palabras clave: difusión de iones de litio, ánodos de aleación, potenciales de aprendizaje automático, aleaciones Li-In y Li-Sn, vías de transporte iónico