Clear Sky Science · en
Structural insights and predictive screening of ion transport in Li-rich alloys via neuroevolution potentials
Why this matters for better batteries
Lithium-ion batteries power our phones, cars, and an increasing share of the electric grid. Yet one stubborn bottleneck remains: how quickly lithium ions can move inside battery electrodes. This paper tackles that problem for a promising class of negative-electrode materials—lithium-rich alloys based on indium and tin—by using advanced machine learning to watch ions move through these complex atomic mazes. The insights could help engineers design batteries that charge faster, last longer, and operate more safely.
Finding the fastest paths through atomic mazes
In alloy anodes, lithium does not merely squeeze between layers; it actually forms new compounds with metals like indium and tin. These alloys can tame some of the safety issues of pure lithium metal, but they introduce a new puzzle: the atomic structures are intricate, and lithium can move in many competing ways. The authors show that three ingredients largely control how fast lithium travels. First is the type of “carrier” that moves: either a missing lithium atom (a vacancy), an extra lithium squeezed between atoms (an interstitial), or a missing metal atom. Second is how well low-resistance routes connect through the crystal. Third is the immediate neighborhood around each moving ion, including how far it must hop and how its electric charge shifts along the way.
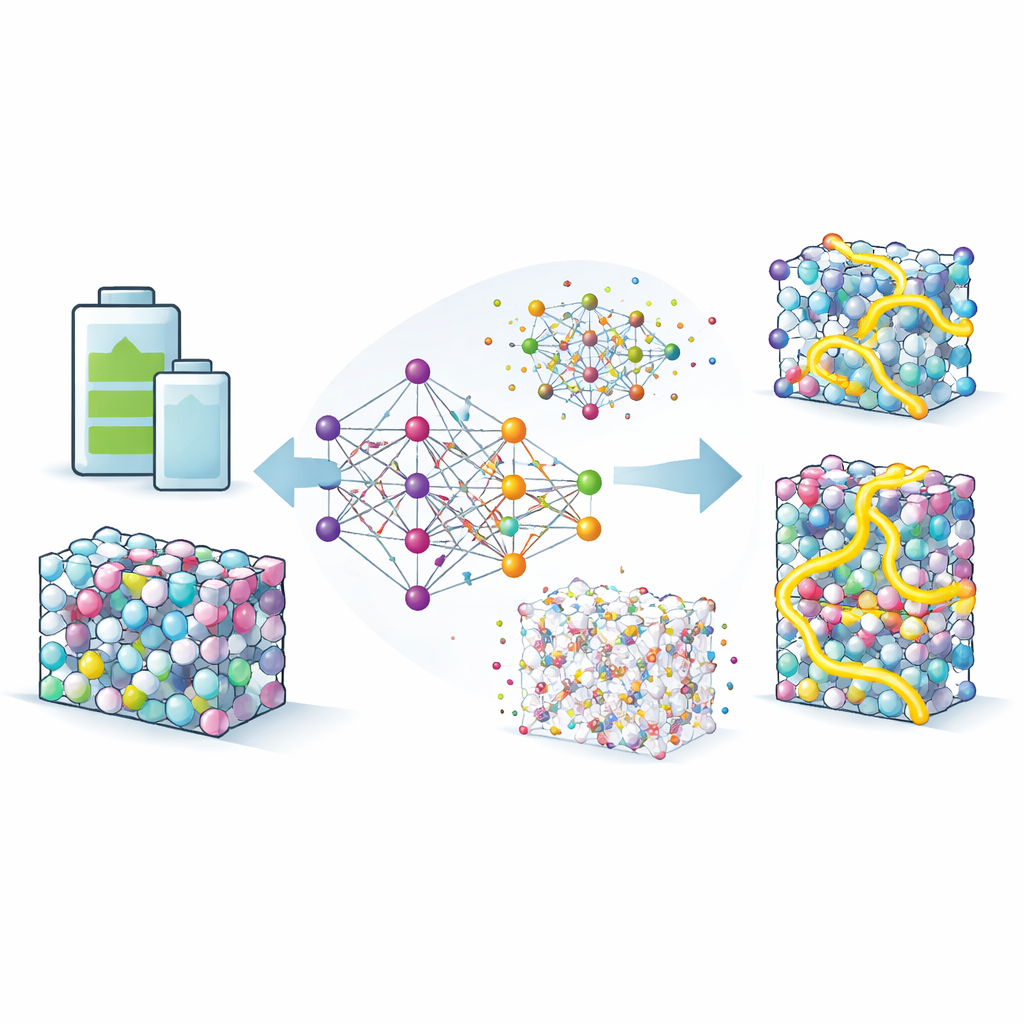
Teaching a digital surrogate for quantum physics
To explore these effects in detail, the team builds so-called neuroevolution potentials, a type of machine-learning model trained to imitate the accuracy of quantum mechanical calculations at a fraction of the cost. They feed this model with thousands of atomic arrangements drawn from databases and from extensive computer searches, then refine it until it reproduces energies and forces with high fidelity. With this surrogate in hand, they can run long and large-scale simulations in which lithium atoms wander through Li–In and Li–Sn alloys at realistic temperatures. The model not only matches experiments on diffusion rates in a benchmark compound, it also correctly reproduces subtle structural properties, giving confidence that it can be trusted to explore unfamiliar phases.
New alloy structures and how they breathe
Armed with the fast surrogate, the researchers systematically search for stable and nearly stable compounds in the lithium–indium and lithium–tin systems. They recover most previously known phases and predict several new ones that appear both energetically and dynamically viable, hinting they could be made in the lab. As more lithium is added, the network of indium or tin atoms evolves from extended three-dimensional frameworks to sheets, chains, and eventually isolated atoms, while lithium donates electrons and takes on a mostly ionic role. The simulations also track how much these alloys swell as they absorb lithium—by about two and a half times in volume at full loading—consistent with experimental observations and crucial for judging mechanical durability in real batteries.
How lithium really moves inside these alloys
By following individual atoms in motion, the study reveals when and how different carriers dominate. At low lithium contents, isolated lithium atoms encourage extra interstitial ions to hop by “knocking off” neighbors in low-energy moves. At higher lithium levels, continuous lithium networks form, and vacancies—missing lithium sites—become the main carriers, diffusing along connected channels. The overall speed, however, is not set just by the lowest single barrier, but by whether many low-barrier hops link up into long-range highways. In some structures, a web of gently sloping routes enables lithium to traverse the crystal with ease; in others, low-resistance segments are trapped in dead ends, and motion slows dramatically. Tin-based alloys largely mirror this behavior, with subtle differences arising from slightly stronger bonding.
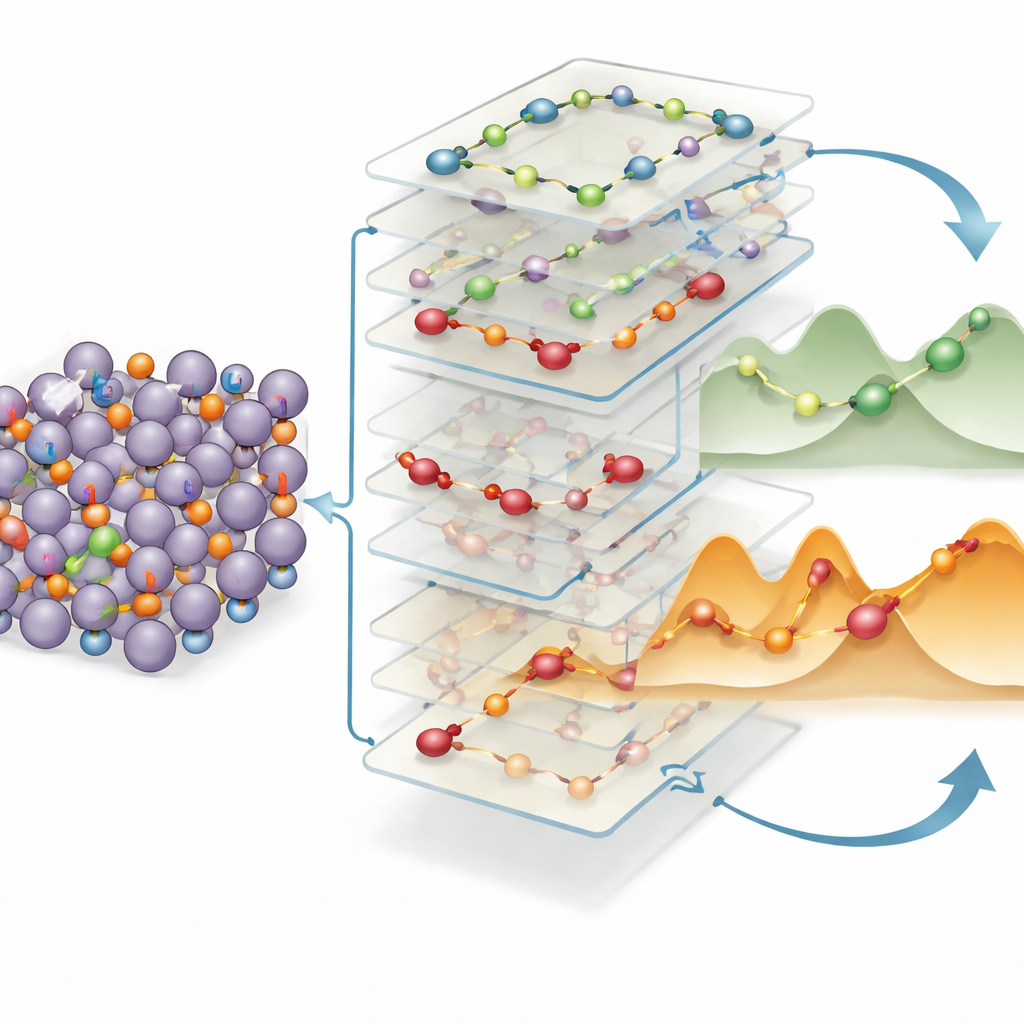
Simple rules for spotting fast ion highways
To turn these insights into practical design rules, the authors train another machine-learning model to relate local structural descriptors—hop distance, how much charge shifts, local bonding geometry, and more—to the energy cost of each move. They find that two factors dominate: how far lithium must jump and how much its charge distribution is disrupted between start and finish. Longer hops and larger charge rearrangements reliably mean higher barriers. Strikingly, the same favorable patterns—short hops along well-connected paths with gentle charge changes—reappear in a wide range of lithium-rich alloys beyond indium and tin, including compounds containing silicon and germanium. This structural “inheritance” suggests that engineers can screen databases of materials for these motifs to rapidly pinpoint new alloy anodes with inherently fast lithium transport.
What this means for future batteries
In everyday terms, this work shows that the best alloy anodes are those whose atomic scaffolding offers lithium a network of short, smoothly graded stepping-stones rather than a landscape of isolated puddles and steep hills. By combining accurate machine-learning surrogates of quantum mechanics with clear structural rules, the study lays out a roadmap for scanning huge families of lithium-rich alloys on a computer before making them in the lab. That could accelerate the discovery of battery electrodes that charge quickly, carry more energy, and cycle reliably over years of use.
Citation: Jin, D., Ding, S., Qiu, H. et al. Structural insights and predictive screening of ion transport in Li-rich alloys via neuroevolution potentials. npj Comput Mater 12, 132 (2026). https://doi.org/10.1038/s41524-026-02012-1
Keywords: lithium-ion diffusion, alloy anodes, machine learning potentials, Li-In and Li-Sn alloys, ion transport pathways