Clear Sky Science · fr
Flux de travail général pour localiser les hydrures dans des nanoclusters métalliques en combinant la marche stochastique de surface et des potentiels par réseau de neurones
Pourquoi les petits clusters métalliques comptent
De nombreuses technologies modernes, des dispositifs d’énergie propre à l’éclairage intelligent, reposent sur des matériaux constitués de clusters d’à peine quelques dizaines d’atomes métalliques. Souvent, ces clusters renferment des atomes d’hydrogène, jouant le rôle de mini-réservoirs ou de sites réactifs. Connaître précisément la position de ces hydrures est essentiel pour comprendre et améliorer le fonctionnement de ces matériaux, mais détecter de si légers atomes dans des structures métalliques denses est extrêmement difficile avec les outils expérimentaux standards.
Trouver des aiguilles d’hydrogène dans une meule de métal
Les atomes d’hydrogène sont presque invisibles aux techniques courantes de rayons X, si bien que les chercheurs se tournent généralement vers les sources de neutrons pour les localiser. Toutefois, les installations neutroniques puissantes sont rares, ce qui limite les études de routine. Des travaux antérieurs ont montré que l’apprentissage profond pouvait deviner les positions d’hydrogène dans certains clusters de cuivre, mais cette approche dépendait de grands jeux de données spécialisés et ne se généralisait pas bien à d’autres métaux. La nouvelle étude introduit un flux de travail informatique largement applicable capable de localiser les hydrures dans de nombreux types de nanoclusters métalliques sans recourir aux données neutroniques, rendant ce type d’analyse plus accessible aux laboratoires du monde entier.
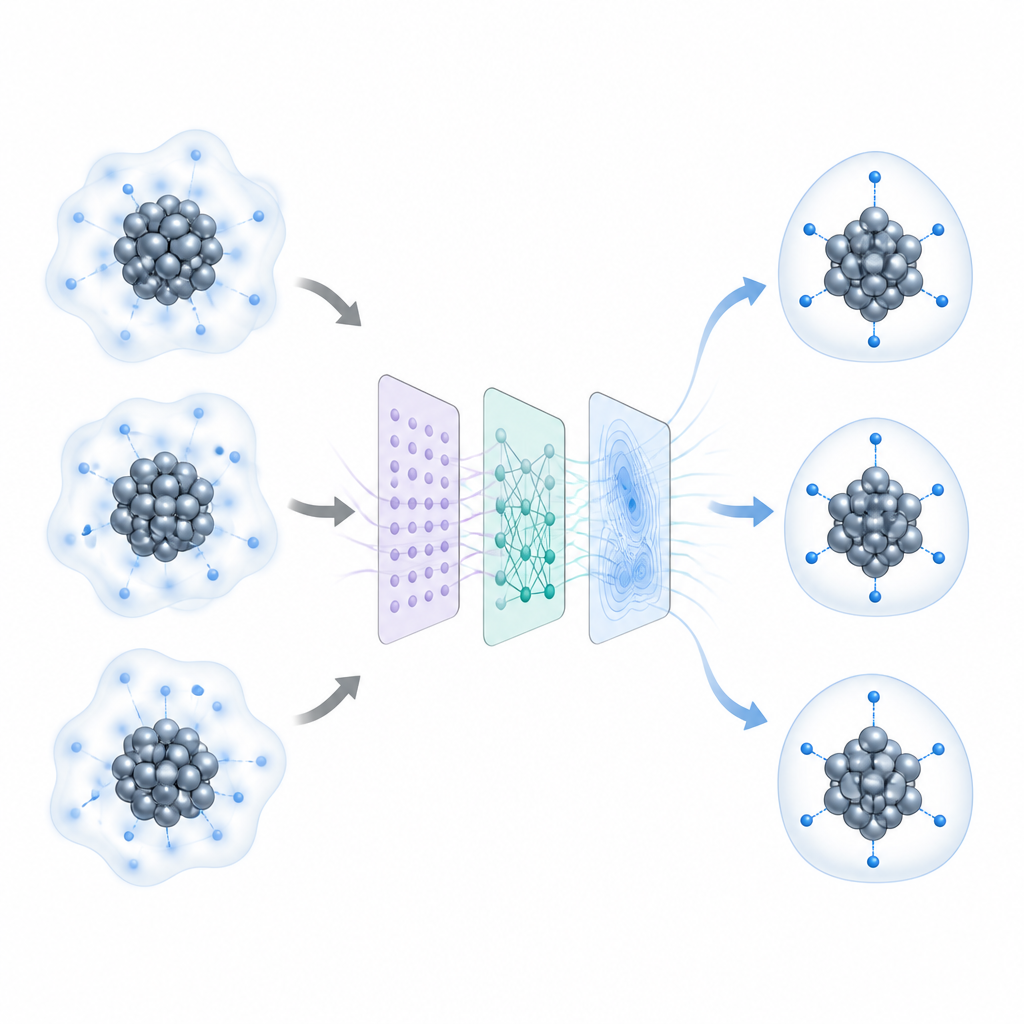
Décomposer les clusters complexes en parties plus simples
Les chercheurs considèrent chaque nanocluster comme la combinaison de trois éléments : un cœur métallique, la couche où le métal rencontre les molécules environnantes, et une coque externe de ligands protecteurs. Ils utilisent une stratégie de recherche globale appelée marche stochastique de surface pour explorer de nombreuses configurations atomiques possibles, tandis que des modèles rapides de réseau de neurones estiment leurs énergies presque aussi précisément que les calculs quantiques, mais bien plus vite. Pour garder le problème gérable et réutilisable, ils simplifient les ligands externes en fragments plus petits qui préservent leur mode de liaison au métal tout en supprimant les détails inutiles. Des tests sur plusieurs clusters riches ou pauvres en hydrogène montrent que cette simplification modifie à peine les positions d’hydrogène prédites, tout en réduisant le temps et le coût de calcul.
Règles sur les endroits où l’hydrogène aime se placer
En appliquant le flux de travail à 93 systèmes publiés, incluant des clusters de cuivre, d’argent, d’or, des alliages et même des composés très différents comme des cages de polyoxométalates, l’équipe cartographie des milliers d’environnements locaux d’hydrogène. Des tendances nettes émergent. Dans les clusters de cuivre, l’hydrogène fait le plus souvent le lien entre trois ou quatre atomes de cuivre, les coordinations plus élevées devenant de plus en plus rares. Les clusters d’argent sont dominés par des sites à trois coordinations, tandis que les clusters d’or accueillent plutôt des hydrures à deux coordinations et occasionnellement à une coordination. Le dopage du cœur métallique par des métaux de transition plus lourds tels que le platine, l’iridium ou le ruthénium favorise fortement la formation de liaisons directes métal–hydrogène, alors que l’échange entre cuivre, argent et or entre eux modifie peu les positions de l’hydrogène tant que la structure globale reste intacte.

Vérifier les formules des clusters et observer le mouvement de l’hydrogène
Comme la spectrométrie de masse et la résonance magnétique nucléaire peuvent mal compter quelques atomes d’hydrogène, les auteurs testent si leur méthode peut aussi aider à confirmer le nombre réel d’hydrogènes présents dans un cluster. Pour un cluster d’or de composition connue, ils effectuent des recherches en supposant un nombre d’hydrogènes trop faible, correct et trop élevé. Seul le nombre correct produit une structure d’énergie basse qui correspond au réseau métallique mesuré et à la symétrie attendue ; des comptes erronés forcent le cluster à se déformer ou à perdre des hydrures. L’équipe va plus loin en utilisant une technique connexe pour suivre comment les hydrures sautent d’un site à l’autre. Ils constatent que les déplacements le long de la surface du cluster requièrent généralement beaucoup moins d’énergie que la traversée de l’intérieur, ce qui suggère que la plupart des échanges s’effectuent par migration de surface.
Ce que cela signifie pour les matériaux futurs
En combinant une recherche globale intelligente avec des potentiels rapides par réseau de neurones, les auteurs proposent une recette pratique pour révéler où se cachent les hydrures dans des nanoclusters complexes et des matériaux apparentés. Pour les non-spécialistes, le message clé est que les ordinateurs peuvent désormais compléter de manière fiable les informations manquantes sur l’hydrogène que les seules expériences ne fournissent souvent pas. Cela facilite l’interprétation des mesures, la conception de nouveaux catalyseurs et la compréhension de la manière dont de petits clusters métalliques stockent et déplacent l’hydrogène, soutenant en fin de compte de meilleurs matériaux pour la catalyse, la conversion d’énergie et la détection chimique.
Citation: Wang, Z., Fang, C., Zhang, L. et al. General workflow for localizing hydrides in metal nanoclusters by combining stochastic surface walking with neural-network potentials. Nat Commun 17, 4513 (2026). https://doi.org/10.1038/s41467-026-72966-9
Mots-clés: nanoclusters métalliques, localisation des hydrures, potentiels par réseau de neurones, marche stochastique de surface, migration de l’hydrogène