Clear Sky Science · de
Allgemeiner Arbeitsablauf zur Lokalisierung von Hydriden in Metallnanoklustern durch Kombination von Stochastic Surface Walking mit neuronalen Netzwerk-Potenzialen
Warum winzige Metallcluster wichtig sind
Viele moderne Technologien, von sauberer Energietechnik bis zu intelligenter Beleuchtung, beruhen auf Materialien, die aus Clustern von nur wenigen Dutzend Metallatomen bestehen. Häufig verbergen diese Cluster Wasserstoffatome in ihrem Inneren und fungieren damit wie winzige Speichertanks oder Reaktionszentren. Zu wissen, wo genau diese Wasserstoffe sitzen, ist entscheidend, um das Verhalten dieser Materialien zu verstehen und zu verbessern, doch solche leichten Atome in dichten Metallstrukturen mit üblichen experimentellen Methoden zu finden, ist extrem schwierig.
Versteckte Wasserstoffnadeln in einem Metall-Heuhaufen finden
Wasserstoffatome sind für gängige Röntgentechniken nahezu unsichtbar, weshalb Forschende in der Regel auf Neutronenquellen zurückgreifen, um sie zu lokalisieren. Leistungsfähige Neutroneneinrichtungen sind jedoch selten, was routinemäßige Untersuchungen einschränkt. Frühere Arbeiten zeigten, dass Deep Learning Wasserstoffpositionen in bestimmten Kupferclustern vorhersagen kann, doch dieser Ansatz war auf große, spezialisierte Trainingsdaten angewiesen und ließ sich kaum auf andere Metalle übertragen. Die neue Studie stellt einen breit einsetzbaren Computer-Workflow vor, der Hydride in vielen Arten von Metallnanoklustern lokalisieren kann, ohne Neutronendaten zu benötigen, und macht diese Art von Analyse so für Labore weltweit zugänglicher.
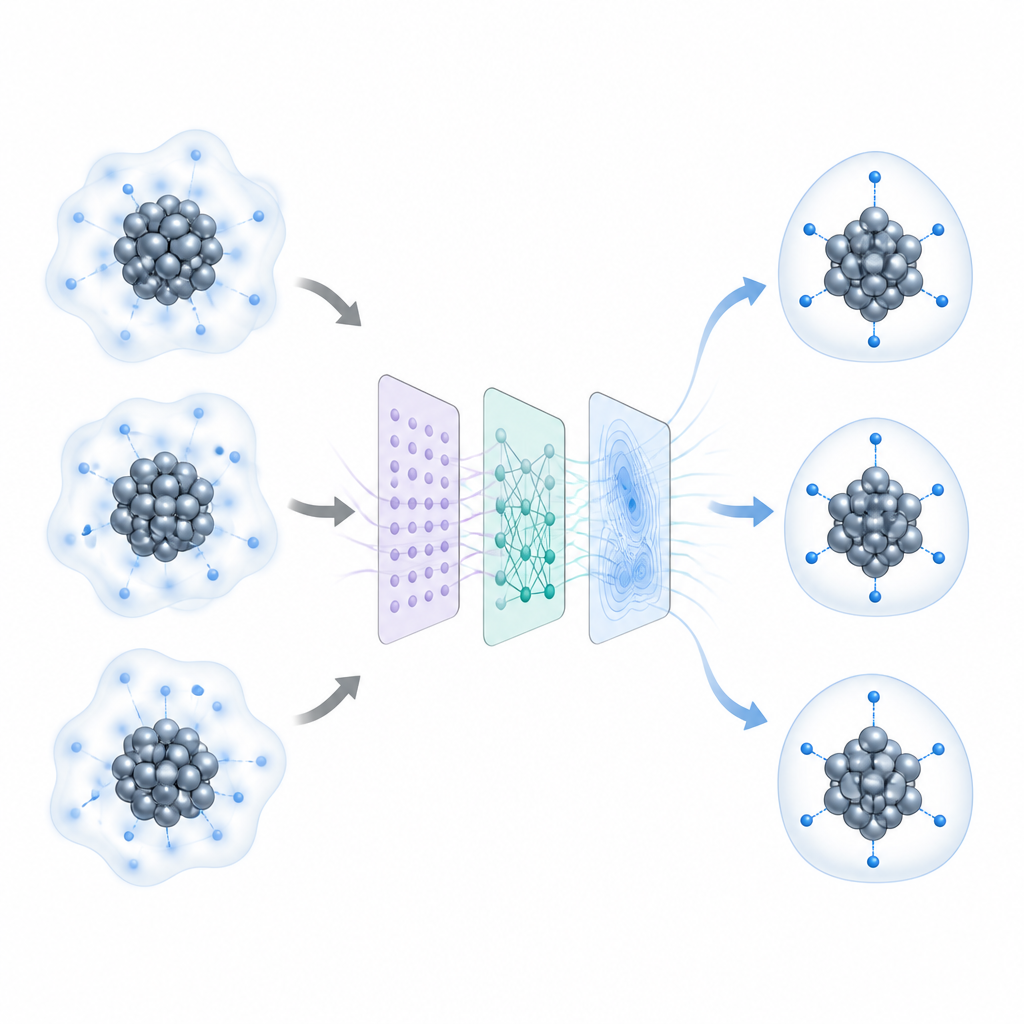
Komplexe Cluster in einfachere Teile zerlegen
Die Forschenden behandeln jeden Nanocluster als Kombination aus drei Teilen: einem metallischen Kern, der Schicht, in der das Metall auf umgebende Moleküle trifft, und einer äußeren Schicht schützender Liganden. Sie nutzen eine globale Suchstrategie namens stochastic surface walking, um viele mögliche atomare Anordnungen zu erkunden, während schnelle neuronale Netzmodelle deren Energien nahezu so genau wie Quantenberechnungen, aber deutlich schneller abschätzen. Um das Problem handhabbar und wiederverwendbar zu machen, vereinfachen sie die äußeren Liganden zu kleineren Fragmenten, die die Bindungsart an das Metall erhalten, aber unnötige Details entfernen. Tests an mehreren hydridreichen und hydridarmen Clustern zeigen, dass diese Vereinfachung die vorhergesagten Wasserstoffpositionen kaum verändert, gleichzeitig aber Rechenzeit und -kosten erheblich reduziert.
Regeln, wo Wasserstoff gerne sitzt
Bei Anwendung des Workflows auf 93 berichtete Systeme, darunter Kupfer-, Silber-, Gold- und Legierungscluster sowie sehr unterschiedliche Verbindungen wie Polyoxometallat-Käfige, kartiert das Team tausende lokale Wasserstoffumgebungen. Es zeigen sich klare Muster. In Kupferclustern überbrückt Wasserstoff am häufigsten drei oder vier Kupferatome, wobei höhere Koordinationen zunehmend seltener werden. Silbercluster sind von dreifachen Bindungsstellen dominiert, während Goldcluster dazu neigen, zweibindige und gelegentlich einbindige Wasserstoffe zu beherbergen. Die Dotierung des Metallkerns mit schwereren Übergangsmetallen wie Platin, Iridium oder Ruthenium fördert stark direkte Metall–Wasserstoff-Bindungen, während der Austausch von Kupfer, Silber und Gold untereinander die Wasserstoffpositionen kaum verschiebt, solange das Gesamtgerüst intakt bleibt.

Formeln der Cluster prüfen und Wasserstoffbewegung beobachten
Da Massenspektrometrie und Kernspinresonanz Wasserstoffe um einige Atome falsch zählen können, prüfen die Autoren, ob ihre Methode auch helfen kann zu bestätigen, wie viele Wasserstoffe ein Cluster tatsächlich enthält. Für einen Goldcluster mit bekannter Zusammensetzung führen sie Suchläufe mit zu wenigen, der korrekten und zu vielen Wasserstoffen durch. Nur die korrekte Anzahl liefert eine energiearme Struktur, die zum gemessenen Metallgerüst und zur erwarteten Symmetrie passt; falsche Angaben zwingen den Cluster zur Verzerrung oder zum Verlust von Wasserstoffen. Das Team geht weiter und nutzt eine verwandte Technik, um zu verfolgen, wie Wasserstoffe zwischen Stellen hüpfen. Sie finden heraus, dass sich die Bewegung entlang der Clusteroberfläche meist mit deutlich geringerem Energieaufwand vollzieht als der Durchgang durch das Innere, was darauf hindeutet, dass die meisten Austausche durch Oberflächenmigration stattfinden.
Was das für zukünftige Materialien bedeutet
Durch die Kombination von intelligenter globaler Suche und schnellen neuronalen Netzwerk-Potenzialen bieten die Autorinnen und Autoren ein praktisches Rezept, um aufzudecken, wo sich Wasserstoffe in komplexen Nanoklustern und verwandten Materialien verbergen. Für Nicht-Spezialisten ist die Kernbotschaft, dass Computer nun zuverlässig die fehlenden Wasserstoffdetails ergänzen können, die Experimente allein oft nicht liefern. Das erleichtert die Interpretation von Messungen, das Design neuer Katalysatoren und das Verständnis, wie kleine Metallcluster Wasserstoff speichern und transportieren, und unterstützt letztlich bessere Materialien für Katalyse, Energieumwandlung und chemische Sensorik.
Zitation: Wang, Z., Fang, C., Zhang, L. et al. General workflow for localizing hydrides in metal nanoclusters by combining stochastic surface walking with neural-network potentials. Nat Commun 17, 4513 (2026). https://doi.org/10.1038/s41467-026-72966-9
Schlüsselwörter: Metallnanokluster, Hydrid-Lokalisierung, neuronale Netzwerk-Potenziale, stochastic surface walking, Wasserstoffmigration