Clear Sky Science · fr
Variantes d’albinisme oculocutané dans 28 familles consanguines et classification fonctionnelle d’une variante pathogène profondément intronique du gène TYR
Pourquoi cette recherche est importante
L’albinisme se manifeste souvent par une peau, des cheveux et des yeux très clairs, mais derrière cette différence visible se posent de nombreuses questions pour les familles concernées : quel type d’albinisme avons‑nous, d’autres organes seront‑ils affectés, et nos enfants peuvent‑ils l’hériter ? Cette étude menée chez des familles pakistanaises éclaire ces questions en révélant quels gènes sont impliqués, la fréquence d’une forme syndromique grave et la façon dont un type de modification de l’ADN jusque‑là méconnu peut perturber la production de pigments. Les résultats affinent le diagnostic, orientent le suivi médical et suggèrent des voies futures pour corriger certains défauts génétiques.
Familles aux racines partagées
Les chercheurs se sont concentrés sur 28 grandes familles pakistanaises où les parents étaient apparentés et plusieurs membres de la famille étaient atteints d’albinisme. De telles familles sont particulièrement utiles en génétique car les personnes affectées ont plus de chances de porter les mêmes altérations héréditaires de l’ADN. Au total, 136 personnes atteintes d’albinisme ont participé. En recueillant soigneusement des informations cliniques et des échantillons sanguins, l’équipe a pu relier des traits visibles comme la couleur de la peau et des cheveux aux causes génétiques sous‑jacentes. Cette approche leur a permis de résoudre le casse‑tête génétique pour chaque famille étudiée, un taux de réussite exceptionnel par rapport à des travaux antérieurs menés principalement dans des populations européennes.
Identifier les gènes responsables
Grâce au séquençage nouvelle génération, l’équipe a examiné chez chaque famille les variations dans 20 gènes connus pour être associés à l’albinisme. Ils ont également recherché des pertes plus larges de segments d’ADN, appelées variants du nombre de copies. La plupart des familles présentaient des altérations dans deux gènes pigmentaires majeurs : TYR, qui donne l’instruction pour fabriquer une enzyme clé de la pigmentation, et OCA2, qui contribue au bon fonctionnement des compartiments cellulaires producteurs de pigments. Ensemble, ces deux gènes expliquent près de quatre familles sur cinq. 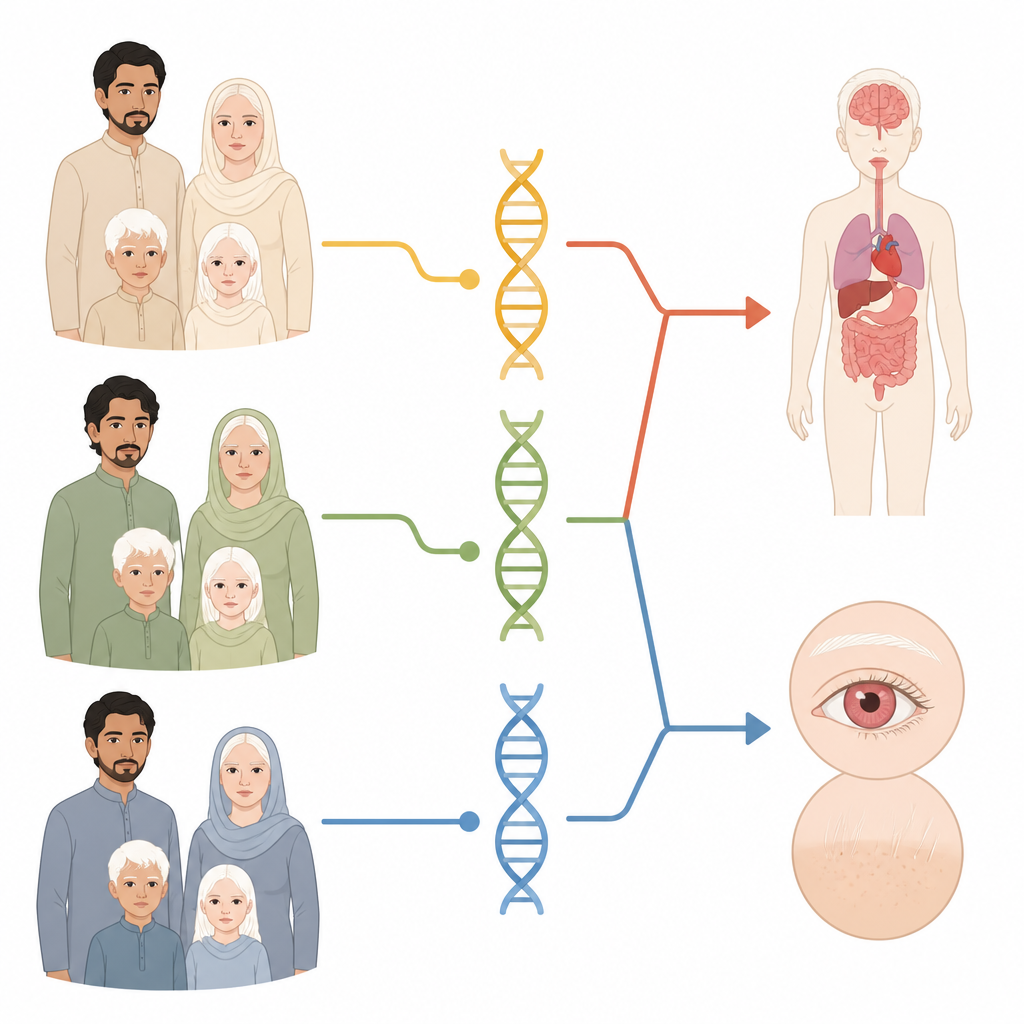
Risques cachés d’une maladie systémique
Tous les albinismes ne se limitent pas aux yeux, aux cheveux et à la peau. Certaines formes, regroupées sous le syndrome d’Hermansky–Pudlak, affectent aussi la coagulation sanguine, l’immunité, les poumons ou les intestins. Dans cette étude, cinq des 28 familles présentaient des altérations délétères dans des gènes liés à ce syndrome. Cela signifie qu’au sein de ce groupe, près d’une famille sur cinq avait en fait une forme syndromique susceptible d’entraîner des complications graves. Parce que la couleur de la peau et des cheveux ne suffit pas à le révéler, les auteurs insistent sur l’importance d’un dépistage génétique étendu incluant ces gènes syndromiques, afin que les cliniciens puissent surveiller les troubles du saignement, les infections et d’autres problèmes.
Une anomalie cachée profondément au sein d’un gène
Dans une famille, aucune modification évidente délétère n’était détectée dans les parties codantes usuelles des gènes pigmentaires, même après un dépistage approfondi. Pour aller plus loin, l’équipe a séquencé le génome complet de plusieurs membres et recherché des segments d’ADN partagés. Cela a pointé vers le gène TYR, mais la variation suspecte se trouvait loin à l’intérieur d’un de ses introns, ces portions d’ADN normalement éliminées lors du traitement des messages génétiques. Des expériences en laboratoire utilisant une version miniature du gène ont montré que cette variante intronique profonde crée un nouveau « pseudoexon », un fragment supplémentaire inséré par erreur dans l’ARN messager. 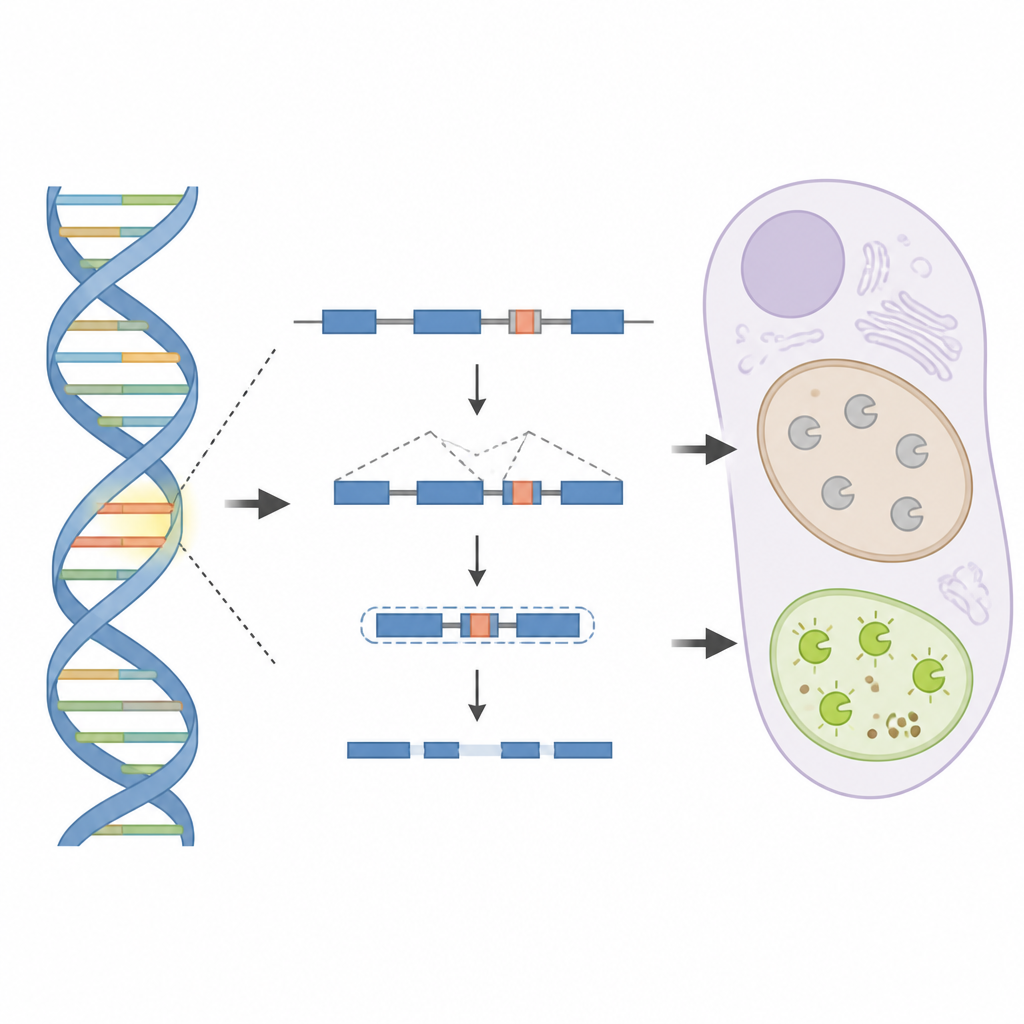
Tester des moyens de réparer les messages génétiques
Fait intéressant, les chercheurs ne se sont pas arrêtés à l’identification du défaut. Ils ont conçu de courts segments d’ARN synthétiques qui se lient aux sites d’épissage fautifs entourant le pseudoexon et en bloquent l’utilisation. Lorsqu’ils sont ajoutés à des cellules portant le mini‑gène mutant, ces molécules modifiant l’épissage ont réduit l’inclusion du fragment supplémentaire et restauré un profil de traitement de l’ARN plus proche de la normale. Bien que ces travaux en soient encore au stade expérimental, ils montrent que certaines erreurs introniques profondes pourraient un jour être partiellement corrigées au niveau de l’ARN, ce qui pourrait améliorer l’activité des enzymes pigmentaires même chez des individus portant la mutation d’ADN sous‑jacente.
Que cela signifie pour les personnes atteintes d’albinisme
Concrètement, cette étude montre qu’un dépistage génétique approfondi peut révéler non seulement quel gène cause l’albinisme d’une personne, mais aussi si elle est à risque de complications au‑delà des changements cutanés et oculaires. Elle démontre également que les variantes pathogènes ne se limitent pas aux régions codantes bien connues des gènes, mais peuvent se cacher profondément dans des régions non codantes qui perturbent subtilement l’assemblage des messages génétiques. En cartographiant ces défauts cachés et en testant des moyens de les contourner, les chercheurs posent les bases de diagnostics plus précis et, à plus long terme, de thérapies ciblées susceptibles d’améliorer la fonction pigmentaire pour certaines formes d’albinisme.
Citation: Farooq, M., Bruun, G.H., Sarusie, M.V.K. et al. Oculocutaneous albinism variants in 28 consanguineous families and functional classification of a pathogenic deep intron variant in TYR. Eur J Hum Genet 34, 603–608 (2026). https://doi.org/10.1038/s41431-026-02070-5
Mots-clés: albinisme oculocutané, gène TYR, syndrome d’Hermansky–Pudlak, pseudoexon, diagnostic génétique