Clear Sky Science · es
Caracterización estructural y funcional in silico de variantes missense de alto riesgo en los genes MMP8, GZMK y OASL asociadas con infecciones virales epidémicas
Por qué pequeños cambios genéticos importan en grandes brotes
Cuando un nuevo virus se difunde por la población, no todos enferman de la misma manera. Algunas personas sortean la infección, mientras que otras desarrollan enfermedades potencialmente fatales. Este estudio plantea una pregunta aparentemente sencilla: ¿pueden pequeños cambios heredados en nuestros genes inmunitarios ayudar a explicar estas diferencias? Usando potentes simulaciones por ordenador en lugar de experimentos de laboratorio, los investigadores exploran cómo variantes genéticas concretas podrían remodelar sutilmente proteínas inmunitarias clave y, a su vez, alterar la respuesta del organismo frente a virus epidémicos como la gripe, el ébola o los coronavirus.
Tres colaboradores del sistema inmunitario bajo el microscopio
El equipo se centró en tres genes humanos que estudios previos a gran escala habían señalado como actores centrales en las respuestas frente a muchos virus respiratorios: MMP8, GZMK y OASL. Cada gen codifica una proteína que contribuye a controlar cómo combatimos la infección. MMP8 ayuda a remodelar tejido dañado y a regular la inflamación en los pulmones y en otros órganos. GZMK codifica la granzima K, una enzima liberada por células asesinas del sistema inmune para atacar células infectadas por virus e influir en la inflamación. OASL produce una proteína que refuerza el sistema de alarma antiviral del organismo y puede interferir directamente con la replicación viral. Dentro de estos genes, los autores identificaron cinco variantes missense raras pero de alto riesgo—cambios de una sola letra en el ADN que sustituyen un aminoácido por otro—porque múltiples herramientas predictivas coincidieron en que probablemente eran dañinas.
Simulando el impacto de variantes dañinas
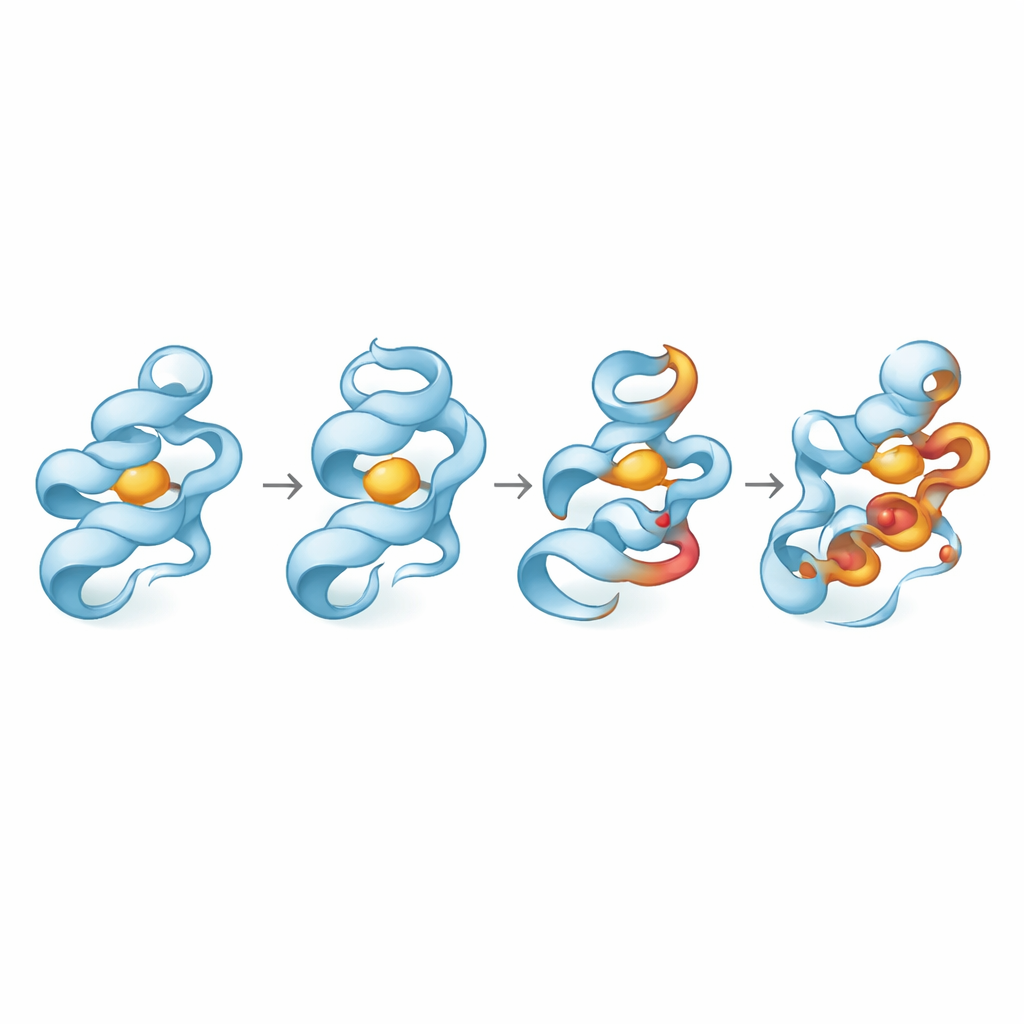
En lugar de probar estas variantes en células o animales, los investigadores construyeron modelos 3D digitales de las proteínas normales y alteradas y los sometieron a una batería de pruebas in silico. Utilizaron algoritmos consolidados para evaluar si cada cambio se predecía como dañino para la proteína, cuánto podría debilitar su estabilidad global y si afectaba regiones altamente conservadas entre especies, indicador de importancia funcional. A continuación, realizaron largas simulaciones de dinámica molecular—esencialmente películas basadas en la física a escala atómica—para observar cómo cada proteína mutante se flexionaba, compactaba o relajaba durante cientos de nanosegundos y cómo estos cambios influían en sus interacciones con una molécula pequeña representativa.
Diferentes genes, destinos estructurales distintos
Los resultados mostraron que no todas las variantes de riesgo se comportan de la misma manera. En la proteína MMP8, una variante, D253N, provocó que la estructura se encogiera y quedara menos expuesta al agua circundante, lo que sugiere un estado más compacto pero desestabilizado que exploró una gama más amplia de conformaciones. Otra variante de MMP8, Y261S, mantuvo el plegamiento y las dimensiones generales cercanas a lo normal e incluso pareció algo más rígida. En GZMK, el cambio A42P tuvo efectos leves, pero L122P aumentó claramente la flexibilidad local y alteró el empaquetamiento de la proteína. En OASL, la variante W216C aflojó la estructura, aumentó la exposición superficial y perturbó movimientos internos de largo alcance, coherente con un plegamiento relajado y menos cohesivo. Los análisis de componentes principales, que condensan movimientos complejos en unos pocos patrones dominantes, confirmaron que D253N, L122P y W216C ampliaron especialmente el rango de conformaciones exploradas.
Cuando el acoplamiento parece correcto pero el comportamiento cambia
Para evaluar si estos cambios estructurales podrían afectar la función, el equipo acopló cada proteína normal y mutante con una molécula pequeña elegida y refinó esos complejos con simulaciones adicionales y cálculos energéticos. Todas las variantes aún lograron unirse a sus ligandos, a veces con cambios modestos en la fuerza de unión predicha. Sin embargo, la forma en que lo hacían a menudo difería: los patrones de contacto entre proteína y ligando cambiaron y, en algunos casos—como en MMP8 D253N—la energía libre total de unión se volvió menos favorable, lo que apunta a interacciones más débiles o menos fiables. De manera intrigante, la variante OASL W216C mostró en sus cálculos una unión más fuerte a la molécula de prueba pese a su desestabilización general, lo que ilustra que una mayor flexibilidad puede, en ocasiones, mejorar el agarre sobre un socio molecular aun cuando socava otros aspectos del comportamiento proteico.
Qué implica esto para futuros brotes
Para un lector no especialista, el mensaje clave es que pequeños cambios heredados en nuestro ADN pueden remodelar sutilmente proteínas inmunitarias importantes sin llegar a romperlas de forma evidente. Estas cinco variantes en MMP8, GZMK y OASL parecen alterar el equilibrio entre estabilidad, movimiento y contactos moleculares de maneras que podrían influir en cómo un individuo responde a una infección viral—modificando la inflamación, la capacidad de eliminar células infectadas o la señalización antiviral. Este trabajo no demuestra que los portadores de estas variantes lo hagan mejor o peor durante epidemias reales; eso requerirá estudios de laboratorio y clínicos cuidadosos. Pero al señalar qué cambios distorsionan con más fuerza la dinámica proteica, este estudio computacional proporciona una lista priorizada de variantes para su prueba experimental y subraya cómo las herramientas modernas de simulación pueden ayudar a desentrañar por qué algunas personas pueden ser más vulnerables cuando el próximo virus se propague.
Cita: Et-tanjaouy, M., Saih, A., Machich, O. et al. In silico structural and functional characterization of high-risk missense variants in MMP8, GZMK, and OASL genes associated with epidemic viral infections. Sci Rep 16, 12973 (2026). https://doi.org/10.1038/s41598-026-40467-w
Palabras clave: susceptibilidad a infecciones virales, variantes de genes inmunitarios, dinámica de la estructura proteica, inmunología computacional, mutación missense