Clear Sky Science · de
In silico strukturelle und funktionelle Charakterisierung von Hochrisiko-Missense-Varianten in den Genen MMP8, GZMK und OASL, die mit epidemischen Virusinfektionen in Verbindung stehen
Warum winzige genetische Veränderungen bei großen Ausbrüchen wichtig sind
Wenn sich ein neues Virus in der Bevölkerung ausbreitet, erkranken nicht alle Menschen gleich schwer. Manche kommen mit einer Infektion gut zurecht, während andere lebensgefährliche Verläufe entwickeln. Diese Studie stellt eine auf den ersten Blick einfache Frage: Können kleine vererbte Veränderungen in unseren Immunitätsgenen dazu beitragen, diese Unterschiede zu erklären? Mithilfe leistungsfähiger Computersimulationen statt Laborversuchen untersuchen die Forschenden, wie bestimmte genetische Varianten Schlüssel-Immunglobuline dezent umformen könnten und damit die Reaktion des Körpers auf epidemische Viren wie Influenza, Ebola oder Coronaviren verändern könnten.
Drei Helfer des Immunsystems unter dem Mikroskop
Das Team konzentrierte sich auf drei menschliche Gene, die frühere groß angelegte Analysen als zentrale Akteure in der Abwehr vieler Atemwegsviren identifiziert hatten: MMP8, GZMK und OASL. Jedes dieser Gene kodiert ein Protein, das mitsteuert, wie wir Infektionen bekämpfen. MMP8 hilft, beschädigtes Gewebe umzubauen und die Entzündungsreaktion in der Lunge und anderswo zu modulieren. GZMK kodiert Granzyme K, ein Enzym, das von killerischen Immunzellen freigesetzt wird, um virusinfizierte Zellen anzugreifen und Entzündungen zu beeinflussen. OASL produziert ein Protein, das das antivirale Alarmsystem des Körpers verstärkt und direkt in die Virusreplikation eingreifen kann. Innerhalb dieser Gene wählten die Autorinnen und Autoren fünf seltene, aber hochriskante „Missense“-Varianten aus — Einzelbuchstabenänderungen in der DNA, die eine Aminosäure im Protein gegen eine andere austauschen —, weil mehrere Vorhersagewerkzeuge übereinstimmend anzeigten, dass sie wahrscheinlich schädlich sind.
Simulation der Auswirkungen schädlicher Varianten
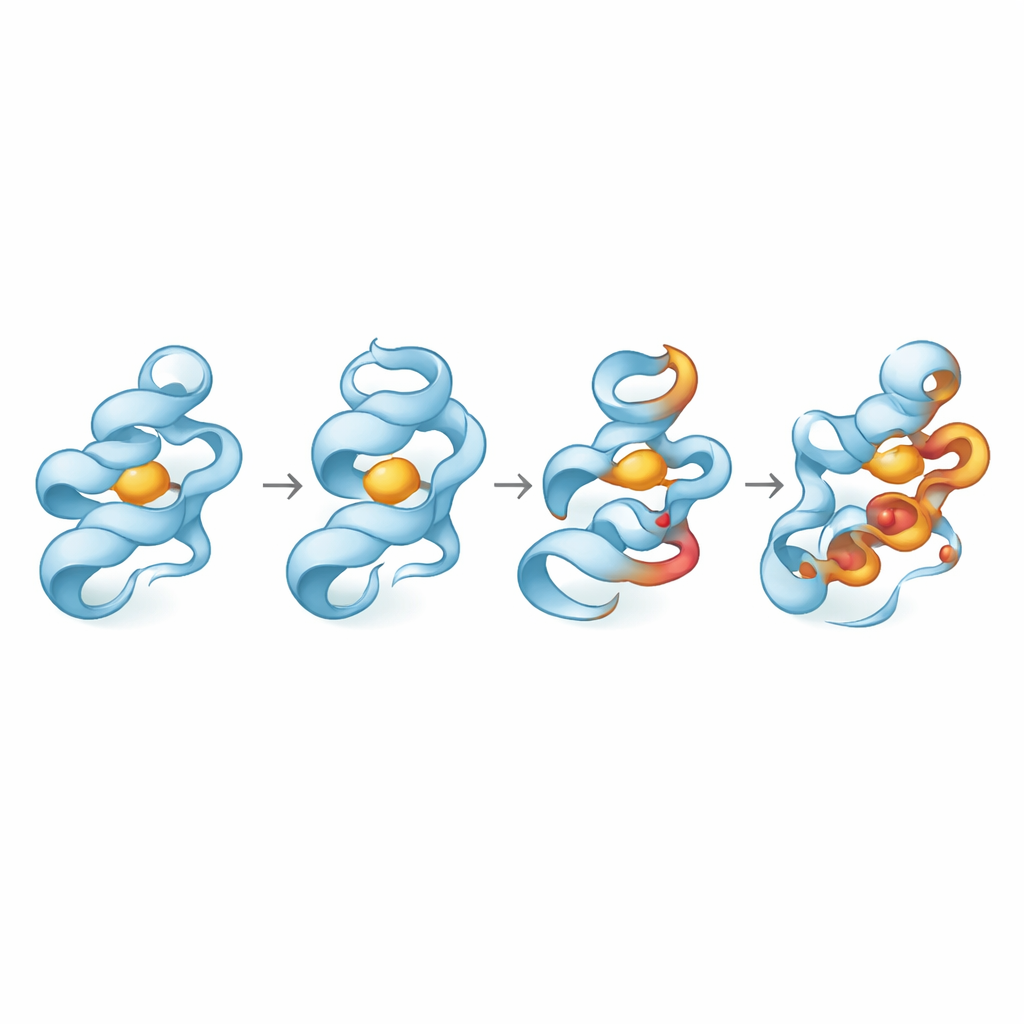
Anstatt diese Varianten in Zellen oder Tieren zu testen, erstellten die Forschenden digitale 3D-Modelle der normalen und veränderten Proteine und unterzogen sie einer Reihe in silico Tests. Sie nutzten etablierte Algorithmen, um zu prüfen, ob jede Änderung das Protein voraussichtlich beschädigt, wie stark sie die Gesamtstabilität schwächen könnte und ob sie Bereiche des Proteins betrifft, die über Arten hinweg hoch konserviert sind — ein Hinweis auf funktionelle Bedeutung. Anschließend führten sie lange Molekulardynamik-Simulationen durch — im Wesentlichen physikbasierte Filme auf atomarer Ebene —, um zu beobachten, wie sich jedes mutierte Protein über hunderte von Nanosekunden verbog, kompaktierte oder lockerte und wie diese Veränderungen seine Wechselwirkungen mit einem repräsentativen kleinmolekularen Partner beeinflussten.
Verschiedene Gene, unterschiedliche strukturelle Schicksale
Die Ergebnisse zeigten, dass nicht alle riskanten Varianten gleich fehlverhalten. Im MMP8-Protein führte eine Variante, D253N, dazu, dass die Struktur schrumpfte und weniger dem umgebenden Wasser ausgesetzt war, was auf einen kompakteren, aber destabilisierteren Zustand hindeutet, der eine größere Bandbreite an Formen durchlief. Eine andere MMP8-Variante, Y261S, bewahrte die Gesamtfaltung und Dimensionen weitgehend und erschien sogar etwas starrer. Bei GZMK hatte die Änderung A42P nur milde Effekte, während L122P deutlich die lokale Flexibilität erhöhte und die Packung des Proteins veränderte. In OASL lockerte die Variante W216C die Struktur, erhöhte die Oberflächenexposition und störte langreichweitige interne Bewegungen, was mit einer entspannten, weniger zusammenhängenden Faltung übereinstimmt. Hauptkomponentenanalysen, die komplexe Bewegungen auf wenige dominante Muster reduzieren, bestätigten, dass D253N, L122P und W216C besonders den Bereich der erkundeten Konformationen vergrößerten.
Wenn die Bindung in Ordnung scheint, sich das Verhalten aber ändert
Um zu prüfen, ob diese strukturellen Verschiebungen funktionell relevant sein könnten, dockte das Team jedes normale und mutierte Protein an ein ausgewähltes Kleinmolekül und verfeinerte diese Komplexe anschließend mit weiteren Simulationen und Energieberechnungen. Alle Varianten gelang es weiterhin, an ihre Partner zu binden, manchmal mit nur mäßigen Veränderungen in der vorhergesagten Bindungsstärke. Dennoch unterschied sich oft die Art und Weise, wie sie banden: Kontaktmuster zwischen Protein und Ligand verschoben sich, und in einigen Fällen — wie bei MMP8 D253N — wurde die gesamte freie Bindungsenergie weniger günstig, was auf schwächere oder weniger verlässliche Wechselwirkungen hindeutet. Interessanterweise zeigte die OASL-Variante W216C trotz ihrer allgemeinen Destabilisierung tatsächlich eine stärkere berechnete Bindung an das Testmolekül, was verdeutlicht, dass erhöhte Flexibilität manchmal den Griff zu einem Partner verbessern kann, während sie andere Aspekte des Proteinverhaltens untergräbt.
Was das für künftige Ausbrüche bedeutet
Für eine nichtwissenschaftliche Leserschaft lautet die Kernaussage, dass winzige vererbte Veränderungen in unserer DNA wichtige Immunproteine dezent umformen können, ohne sie offensichtlich zu zerstören. Diese fünf Varianten in MMP8, GZMK und OASL scheinen das Gleichgewicht zwischen Stabilität, Bewegung und molekularen Kontakten so zu stören, dass sie die individuelle Reaktion auf eine Virusinfektion beeinflussen könnten — etwa durch Änderung von Entzündungsreaktionen, Zelltötung oder antiviraler Signalgebung. Die Arbeit beweist nicht, dass Träger dieser Varianten in realen Epidemien besser oder schlechter abschneiden; dafür sind sorgfältige Labor- und klinische Studien nötig. Aber indem sie herausstellt, welche Änderungen die Proteindynamik am stärksten verzerren, liefert diese computergestützte Studie eine Prioritätenliste von Varianten für experimentelle Prüfungen und unterstreicht, wie moderne Simulationstools helfen können, zu erklären, warum manche Menschen bei der nächsten Viruswelle anfälliger sein könnten.
Zitation: Et-tanjaouy, M., Saih, A., Machich, O. et al. In silico structural and functional characterization of high-risk missense variants in MMP8, GZMK, and OASL genes associated with epidemic viral infections. Sci Rep 16, 12973 (2026). https://doi.org/10.1038/s41598-026-40467-w
Schlüsselwörter: Anfälligkeit für Virusinfektionen, Varianten von Immunitätsgenen, Proteinstrukturdynamik, computationale Immunologie, Missense-Mutation