Clear Sky Science · es
Análisis de datos proteómicos cuantitativos isobáricos usando TMT-Integrator y la plataforma computacional FragPipe
Convertir mediciones proteicas en imágenes claras
La biología moderna suele necesitar comparar miles de proteínas entre numerosas muestras de pacientes, tipos celulares o tratamientos. Hacer esto con los espectrómetros de masas actuales genera océanos de datos que son potentes pero difíciles de domar. Este artículo presenta TMT-Integrator, una herramienta de software integrada en la plataforma FragPipe, que ayuda a los científicos a transformar medidas proteicas complejas en tablas fiables y fáciles de analizar. Al limpiar estos datos y hacerlos más comparables entre experimentos, el trabajo ayuda a los investigadores a comprender mejor enfermedades como el cáncer, probar nuevas tecnologías y combinar datos proteicos con otras lecturas moleculares como el ARN.
Medir muchas muestras a la vez
El estudio se centra en una familia de etiquetas químicas llamadas isobáricas, comúnmente conocidas como TMT e iTRAQ. Estas etiquetas permiten a los científicos mezclar muchas muestras diferentes y analizarlas en una sola corrida en un espectrómetro de masas, acelerando enormemente los experimentos y reduciendo la variación técnica. Cada muestra recibe una etiqueta ligeramente diferente y, cuando las proteínas se fragmentan dentro del instrumento, las etiquetas generan señales distintas que revelan cuánto de cada proteína provino de cada muestra. Este “multiplexado” es hoy rutinario en grandes estudios de cáncer, en el descubrimiento de dianas farmacológicas e incluso en la proteómica de célula única. Sin embargo, las señales crudas están afectadas por ruido, interferencias y diferencias entre lotes de muestras, por lo que se necesita computación sofisticada para interpretar los resultados.
De señales crudas a números fiables
TMT-Integrator se sitúa al final de un flujo de trabajo más amplio en FragPipe que comienza identificando péptidos a partir de espectros de masas y asignándolos a proteínas y genes. La herramienta toma tablas detalladas de coincidencias péptido-espectro y señales de iones reporteros de una o varias corridas multiplexadas y luego realiza una serie de pasos cuidadosamente diseñados. Filtra mediciones poco fiables, convierte intensidades crudas en ratios relativos a una muestra de referencia elegida, agrupa las medidas de péptidos en resúmenes de nivel superior, elimina valores atípicos y después convierte los ratios de nuevo en valores de “abundancia” similares a intensidades. Este proceso puede aplicarse en varios niveles, incluidos genes, proteínas, secuencias de péptidos y sitios específicos de modificación como la fosforilación. TMT-Integrator también ofrece opciones flexibles de normalización y puede trabajar con una muestra de referencia real que aparezca en cada lote o con una referencia “virtual” calculada a partir de los datos.
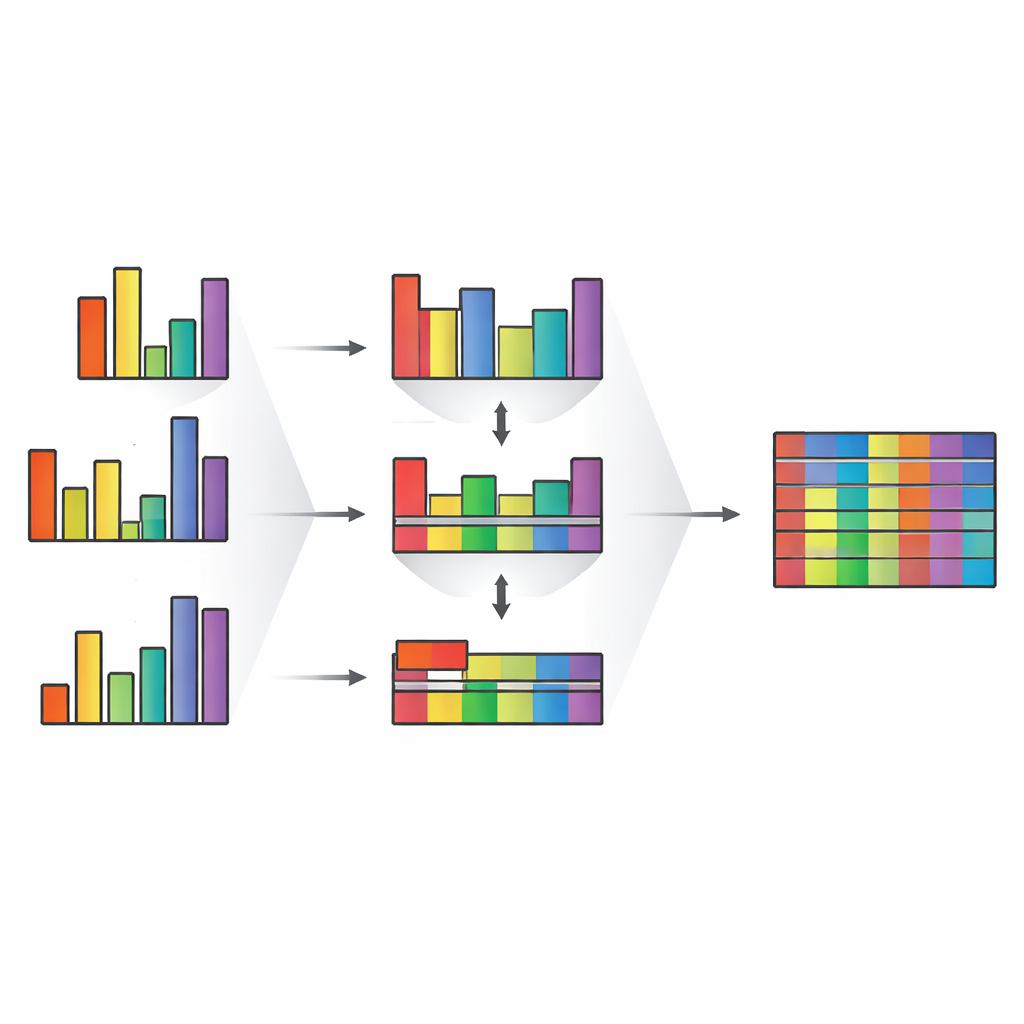
Manejar muchos lotes y muchas tecnologías
Un desafío importante en proyectos grandes es que un conjunto de etiquetas no puede cubrir todas las muestras, por lo que los estudios se dividen en múltiples lotes, o “plexes”. Pequeñas diferencias entre estos plexes pueden ocultar cambios biológicos reales. Los autores muestran cómo la estrategia de ratio-a-referencia de TMT-Integrator, combinada con canales de referencia reales o virtuales, conecta efectivamente estos plexes a la vez que reduce los efectos de lote. Usando conjuntos de datos de carcinoma de células claras del riñón de un programa nacional contra el cáncer, demuestran que FragPipe con TMT-Integrator detecta más proteínas y más sitios de fosforilación que alternativas populares como MaxQuant y Proteome Discoverer, especialmente para proteínas de baja abundancia. Las mediciones proteicas resultantes se alinean mejor con datos de ARN correspondientes, capturan relaciones conocidas dentro de complejos proteicos y vías, y muestran menos ruido en muestras de control de calidad.
Acercándose a las modificaciones de las proteínas
Muchos eventos de señalización en las células dependen de pequeñas marcas químicas añadidas en posiciones específicas de las proteínas. Capturar estas “modificaciones postraduccionales” a nivel de sitio único es técnicamente exigente. TMT-Integrator utiliza información de localización de sitios procedente de una herramienta dedicada para construir vistas tanto de múltiples sitios como de sitios únicos de péptidos modificados. En datos de fosforilación de cáncer renal, ofrece una cobertura extensa de proteínas modificadas, péptidos y sitios individuales, separando tumor y tejido normal con claridad y proporcionando mediciones consistentes entre controles replicados. En comparación con MaxQuant, informa más sitios con buena completitud y mejor concordancia entre corridas repetidas, y produce tablas de estilo abundancia que son más fáciles de integrar con otras capas ómicas.
Pensado para instrumentos de nueva generación
Los autores también prueban TMT-Integrator en conjuntos de datos de vanguardia. En un nuevo espectrómetro Orbitrap Astral, comparan el marcado isobárico con otro enfoque llamado adquisición independiente de datos y encuentran que la canalización TMT en FragPipe logra una cobertura profunda con alta precisión y fuerte concordancia con el método alternativo. En un conjunto de datos separado que usa reactivos de 35-plex que combinan etiquetas normales y etiquetadas con deuterio, la estrategia de referencia virtual del software maneja con éxito cambios sutiles en el tiempo de retención y proporciona cambios de plegamiento proteico consistentes entre líneas celulares, sin necesidad de muestras puente especiales en cada corrida. Estos resultados sugieren que la herramienta puede mantenerse al ritmo del hardware y las químicas de marcado que evolucionan rápidamente.
Qué significa esto para la investigación biológica
En conjunto, el trabajo muestra que TMT-Integrator, como parte de FragPipe, puede convertir experimentos proteómicos multiplexados y complejos en tablas precisas e interpretable en múltiples niveles biológicos. Al mejorar la sensibilidad, reducir artefactos técnicos y proporcionar salidas tanto en formato de ratio como de abundancia, ayuda a los investigadores a establecer conexiones más seguras entre proteínas, genes y estados de enfermedad. Para un lector no especializado, el mensaje clave es que un mejor software para gestionar datos de espectrometría de masas se traduce directamente en historias biológicas más claras, biomarcadores más fiables y una base más sólida para la medicina de precisión.
Cita: Chang, HY., Deng, Y., Li, R. et al. Analysis of isobaric quantitative proteomic data using TMT-Integrator and FragPipe computational platform. Nat Commun 17, 4010 (2026). https://doi.org/10.1038/s41467-026-70118-7
Palabras clave: proteómica cuantitativa, espectrometría de masas, marcado isobárico, canalizaciones de análisis de datos, fosforilación de proteínas