Clear Sky Science · en
Impaired cohesin loading disrupts pancreatic differentiation by Polycomb-driven chromatin rewiring and loop collapse
How Cells Fold Their DNA to Become Pancreas Cells
Inside every cell, two meters of DNA must be folded and refolded so that the right genes turn on at the right time. This paper explores how that folding process helps human stem cells become insulin-producing pancreatic cells—and what happens when a key folding helper goes missing. Because errors in this system are linked to developmental disorders and possibly diabetes, understanding this invisible choreography of DNA has broad implications for health.
The Cell’s DNA-Looping Machine
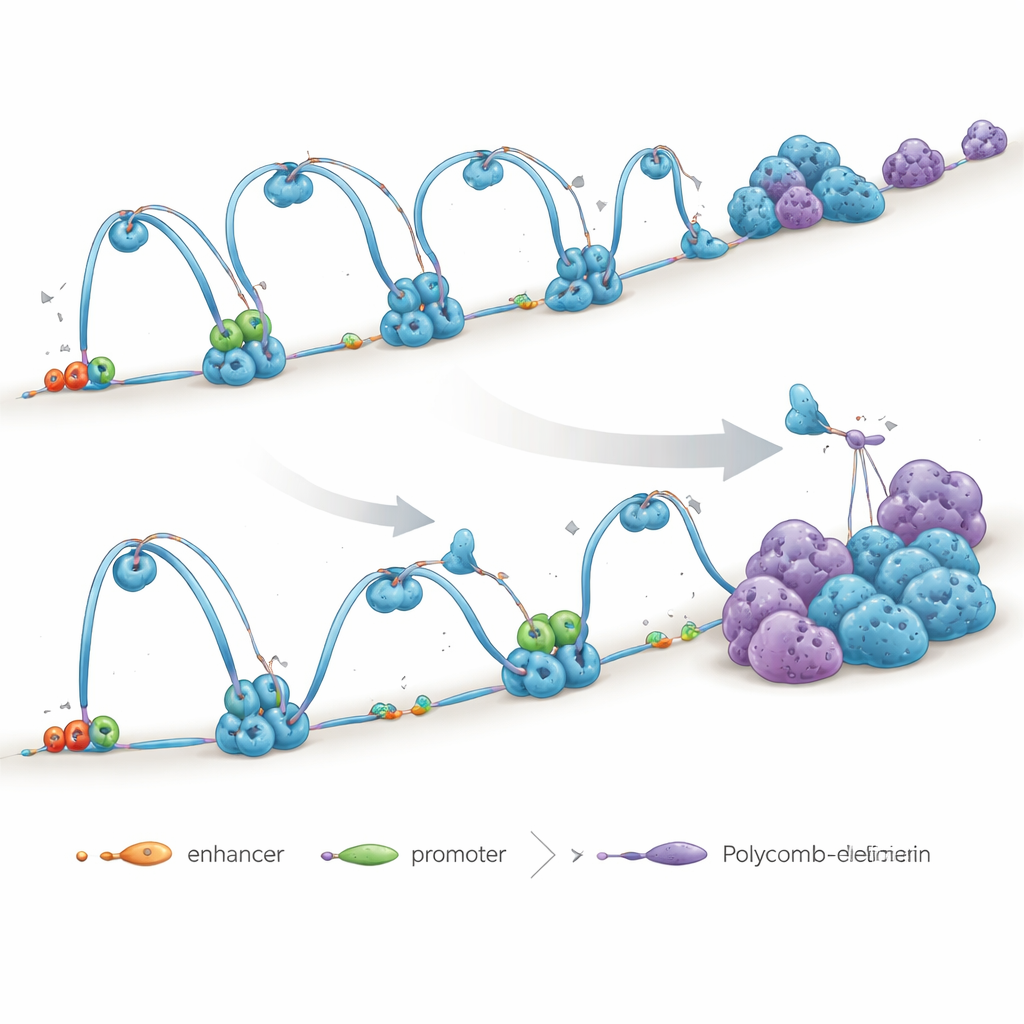
A major focus of the study is a protein helper called NIPBL, which loads a ring-shaped complex known as cohesin onto DNA. Cohesin acts like a sliding clamp that pulls distant stretches of the genome together into loops, bringing gene switches (enhancers) into contact with the genes they control (promoters). Another protein, CTCF, often marks the boundaries of these loops, helping to carve the genome into insulated neighborhoods. The authors show that NIPBL is crucial not only for getting cohesin onto DNA but also for driving the continuous “loop extrusion” that keeps these neighborhoods intact.
What Happens When the Loader Fails
To see what NIPBL really does, the researchers reduced its levels in human embryonic stem cells. Surprisingly, the overall amount of cohesin at many boundary sites stayed high, yet the long-range loops connecting distant segments of DNA weakened or vanished. Enhancer–promoter contacts, which are essential for turning genes on, were especially affected. Even where cohesin remained in place, loops became shorter and less effective, and some large loops appeared to collapse into smaller, local structures. This shows that simply having cohesin present is not enough—its correct loading and movement along DNA, driven by NIPBL, are what sustain the genome’s 3D wiring.
Polycomb Clusters Take Over
DNA is also organized by silencing complexes called Polycomb, which gather groups of genes into repressed compartments. When cohesin-driven loops weakened after NIPBL loss, the authors saw the opposite behavior from Polycomb domains: they interacted more strongly and formed brighter, denser foci inside the nucleus. Long-range contacts between Polycomb-rich regions increased, even though the total amount of Polycomb on DNA changed little. Chemical treatments that disturb droplet-like condensates or interfere with Polycomb’s ability to “read” specific chemical marks reduced these contacts, indicating that Polycomb uses a clustering, phase-separation-like mechanism that becomes more dominant when loop extrusion is impaired. In other words, when loop-based organization fades, compartment-based silencing steps in.
Disrupted Routes to Pancreatic Identity
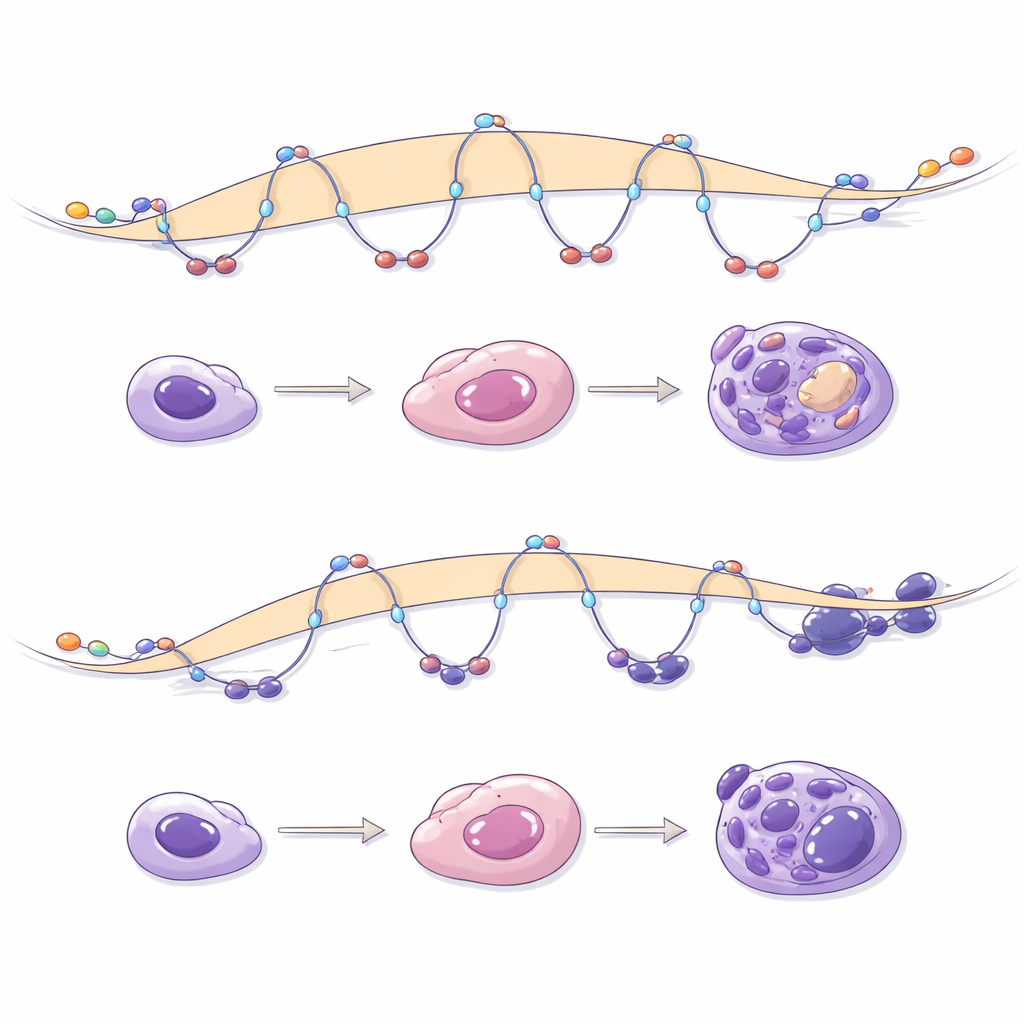
The team then followed stem cells as they were coaxed through several stages toward becoming pancreatic islet organoids, including insulin-producing beta-like cells. Normally, this process involves opening up stage-specific regulatory DNA and building new loops that connect these regions to pancreatic genes. When NIPBL was reduced during differentiation, thousands of genes failed to reach their proper activity levels, and many DNA sites that should have become accessible stayed closed. Newly formed loops that are typical of pancreatic progenitors were fewer, shorter, and weaker, especially those enclosing enhancers and key developmental genes. Inhibiting Polycomb could partially restore gene activity but could not rebuild the missing loops, underscoring that NIPBL-dependent loop formation is a distinct and irreplaceable layer of control.
Super-Switches and Collapsed Circuits
The authors also examined super-enhancers—large clusters of regulatory elements that act as “super-switches” for genes defining cell identity. In normal cells, these regions are knit together and linked to their target genes by cohesin-mediated loops. After NIPBL loss, cohesin marks at many of these super-enhancers declined and the loops connecting them weakened, while some long-distance contacts between different super-enhancer regions increased in a disorganized way. This suggests that when the usual looping circuits break down, the genome may compensate by forming broader, less precise connections, potentially blurring the sharp on/off control needed for developmental genes.
Why This Matters for Development and Disease
Overall, the study paints NIPBL as a master organizer of the 3D genome during cell fate decisions. By loading and mobilizing cohesin, it builds and maintains the loops that let enhancers and promoters communicate, while at the same time counterbalancing the clustering of Polycomb-silenced regions. When NIPBL function is impaired, enhancer–promoter loops collapse, Polycomb compartments tighten, and the carefully timed gene programs needed for pancreatic differentiation falter. This mechanistic picture helps explain how mutations in cohesin-related genes can cause complex developmental syndromes and offers clues to how subtle disruptions in genome folding might contribute to diseases such as diabetes.
Citation: Yu, L., Liu, Y., Zhang, J. et al. Impaired cohesin loading disrupts pancreatic differentiation by Polycomb-driven chromatin rewiring and loop collapse. Commun Biol 9, 590 (2026). https://doi.org/10.1038/s42003-026-09838-x
Keywords: 3D genome organization, cohesin and NIPBL, Polycomb domains, pancreatic differentiation, enhancer–promoter loops