Clear Sky Science · en
Aberrant activation of epigenetic BRD9-DGAT1 axis promotes lipid droplets deposition and ferroptosis resistance in YAP-high prostate cancer
Why this prostate cancer study matters
Many men with advanced prostate cancer eventually face tumors that stop responding to standard hormone therapies. This study uncovers a hidden survival trick inside some of these cancers: they rewire the way they handle fats, helping tumor cells stockpile energy-rich droplets and dodge a type of cell death driven by iron and oxidation. Understanding this wiring offers a new angle for treatment, especially for patients whose tumors show high activity of a growth switch called YAP. 
A growth switch teams up with an epigenetic reader
The researchers focused on a family of protein machines that control how DNA is packaged and read inside cells. Using a genetic screening approach in prostate cancer cells, they pinpointed one member, BRD9, as essential for tumor growth. When BRD9 was blocked or removed, cancer cells grew more slowly, formed fewer colonies, and migrated less in lab tests. In mice, tumors with extra BRD9 grew faster, spread more readily, and shortened survival, while BRD9 loss curbed growth and metastasis. Patient samples showed that BRD9 levels were higher in tumors than in normal prostate tissue and rose with more aggressive disease, linking this protein to poor outcomes.
From YAP activity to BRD9 activation
Next, the team asked what turns BRD9 on in the first place. They tested drugs that shut down several well-known cancer pathways and found that blocking YAP, a key controller of tissue growth, strongly reduced BRD9. Experiments showed that the YAP protein, together with its partner TEAD4, binds directly to the BRD9 control region in DNA and boosts its production. When YAP was removed, prostate cancer cells grew poorly, but restoring BRD9 largely rescued their growth, self-renewal, and migration. In organoids grown from patient tumors, high YAP activity made growth strongly dependent on BRD9, confirming that YAP helps drive malignancy at least in part by switching on this epigenetic reader.
Rewiring fat handling and building protective droplets
To understand what BRD9 does inside the cell, the scientists compared gene activity profiles with and without BRD9. Genes involved in fat and cholesterol metabolism were strongly affected, with one enzyme, DGAT1, standing out. DGAT1 helps convert fatty acids into triacylglycerols that are stored in lipid droplets. BRD9 physically interacted with another regulator, SREBP1, and together they sat at the DGAT1 promoter region, opened up local DNA packaging, and recruited the transcription machinery, all of which boosted DGAT1 levels. When BRD9 or DGAT1 was reduced, prostate cancer cells formed far fewer lipid droplets, while overactive YAP and BRD9 increased droplet buildup. Chemical inhibitors of BRD9 lowered DGAT1 expression and cut down droplet formation, linking this epigenetic circuit directly to the cell’s fat storage compartments. 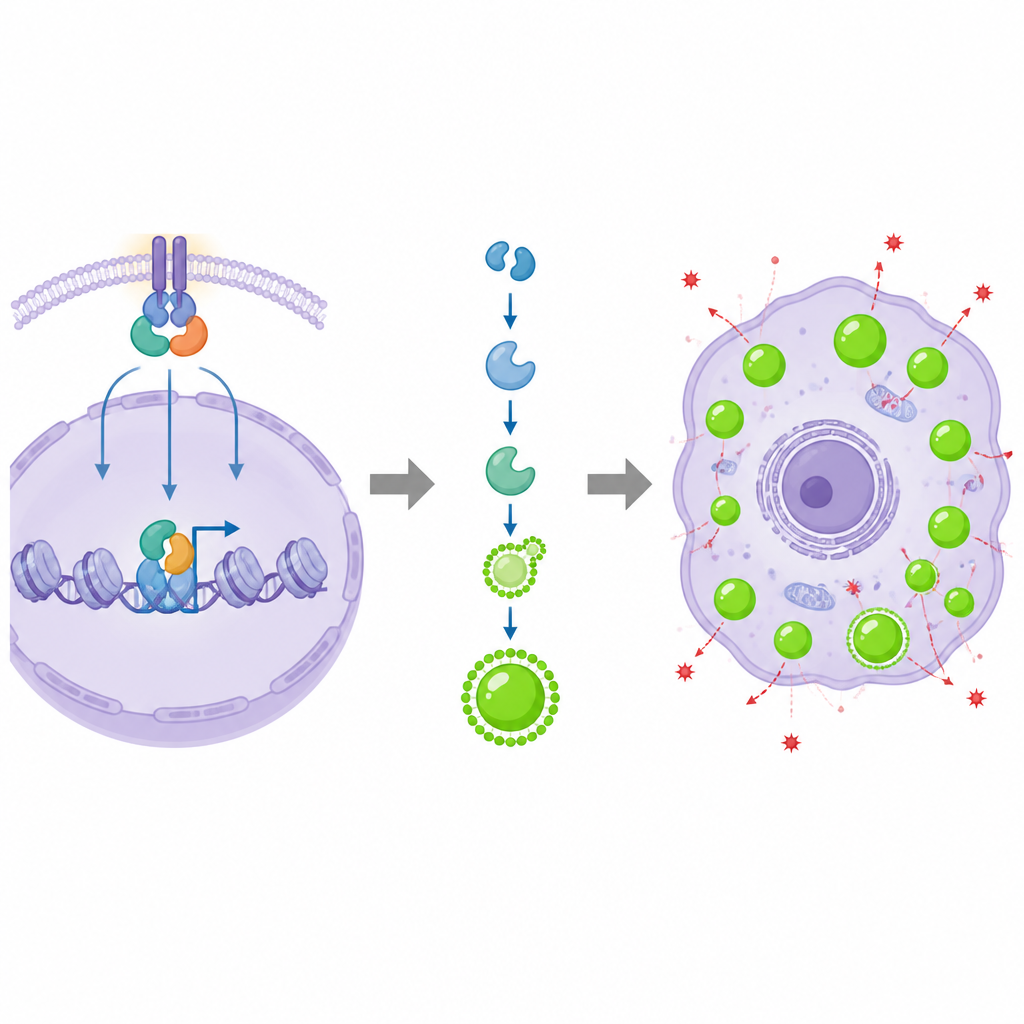
Using fat stores to evade iron-driven cell death
Lipid droplets are more than passive storage; they can shelter vulnerable fats from damage. The team explored ferroptosis, a form of cell death triggered when iron and reactive molecules attack certain fatty acids. Prostate cancer cells lacking BRD9 were much more sensitive to the ferroptosis inducer erastin, an effect that could be reversed by reintroducing normal BRD9 but not a mutant missing its key binding pocket. Boosting DGAT1 rescued cells from death when BRD9 or YAP was blocked, while inhibiting DGAT1 had the opposite effect. The data support a model in which the YAP–BRD9–SREBP1 pathway raises DGAT1 activity, builds up lipid droplets, and tucks away fragile fatty acids, thereby shielding cancer cells from ferroptosis and helping them survive stress.
Targeting the BRD9–DGAT1 pathway in YAP-high tumors
Finally, the researchers tested whether this pathway could be drugged. A small molecule called BI-9564, which targets the bromodomain pocket of BRD9, selectively slowed the growth of prostate cancer cell lines and organoids with high BRD9 and YAP activity, while sparing cells with low BRD9. BI-9564 reduced lipid droplets and made cells more vulnerable to erastin, both in dishes and in mouse tumors. Another drug against DGAT1 showed a similar pattern. In patient-derived tumor grafts, those with high YAP activity and elevated BRD9–DGAT1 responded much better to BRD9 or DGAT1 inhibition than YAP-low tumors, suggesting a way to match therapies to tumor biology.
What this means for future prostate cancer care
This work reveals that some aggressive prostate cancers rely on a YAP-driven BRD9–DGAT1 circuit to reshape how they store fats and resist a lethal, iron-linked form of stress. By disrupting this circuit, either at the epigenetic level with BRD9 inhibitors or at the metabolic level with DGAT1 blockers, it may be possible to weaken tumors that otherwise withstand current treatments. While more safety and clinical studies are needed, the findings highlight a promising strategy: identify patients with “YAP-high” disease and target the fat-storing, ferroptosis-resistant machinery their tumors depend on.
Citation: Zhu, X., Wen, Z., Wu, J. et al. Aberrant activation of epigenetic BRD9-DGAT1 axis promotes lipid droplets deposition and ferroptosis resistance in YAP-high prostate cancer. Cell Death Dis 17, 477 (2026). https://doi.org/10.1038/s41419-026-08746-6
Keywords: prostate cancer, lipid droplets, ferroptosis, BRD9, YAP signaling