Clear Sky Science · en
REP-1 deficiency induces aberrant mitochondrial metabolic rewiring from glycolysis to lipid oxidation in CHM disease
Why this eye research matters
Choroideremia is a rare inherited eye disease that slowly robs people of their sight, often starting with night blindness in childhood and progressing to complete vision loss. This study uncovers how a single missing protein in retinal cells throws their energy balance off, forcing them to burn fats instead of sugar. That shift quietly damages the cells that keep our vision sharp and may point to a new way to treat this and related eye conditions.
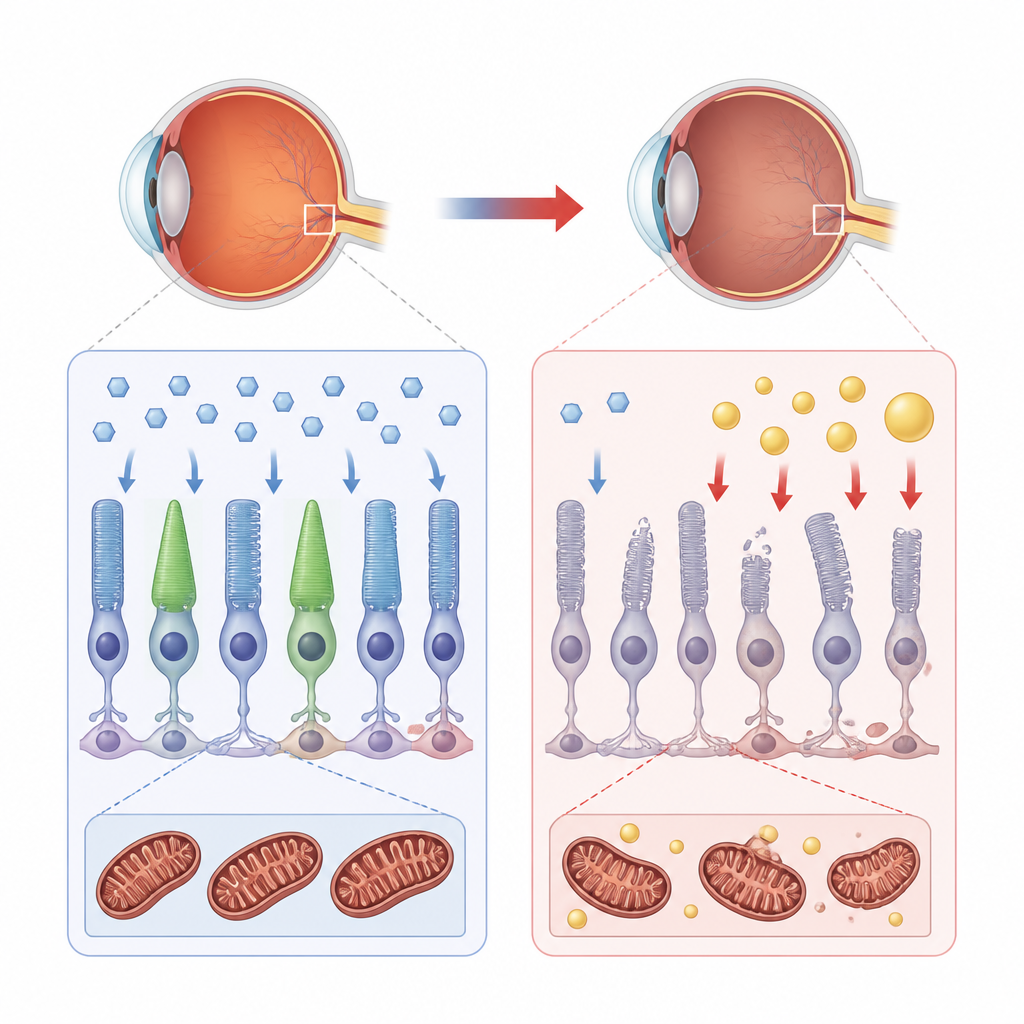
How healthy retinal cells fuel themselves
The retina is one of the most energy-hungry tissues in the body. Under normal conditions, its support cells and light-sensing cells rely mainly on glucose, a simple sugar carried into the cell by special doorway proteins called transporters. Once inside, glucose is broken down through glycolysis and then fed into mitochondria, the tiny power stations that make ATP, the cell’s energy currency. Fats can serve as a backup fuel, but in a healthy retina they play a supporting role rather than the main act.
The missing helper that derails sugar use
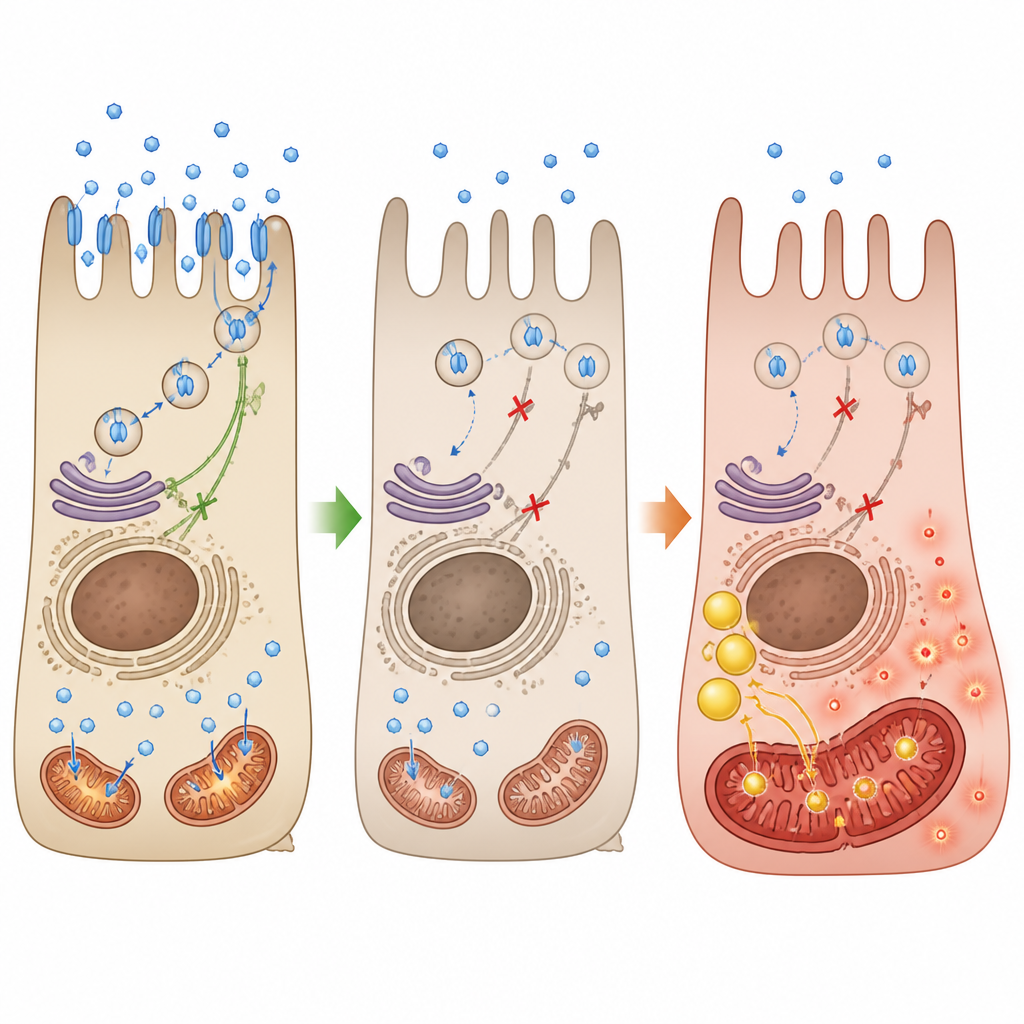
In choroideremia, mutations in the CHM gene remove a protein called REP-1, which normally helps traffic many cargoes inside the cell. The authors used human retinal pigment cells and a small fish model to see what happens when REP-1 is lost. They found that key glucose transporters, GLUT1 and GLUT4, either become less abundant or fail to reach the cell surface. As a result, glucose uptake drops and insulin-like signals that would normally promote transporter movement to the membrane become blunted. Without enough sugar getting in, the cell’s preferred energy route stalls.
When power stations switch from sugar to fat
With glycolysis crippled, the retinal cells shift toward burning lipids for fuel. The team saw an increase in genes linked to lipid metabolism, accumulation of lipid droplets inside cells, and higher levels of oxidized fats. Mitochondria shrank in size, lost their normal branched network, and showed damaged internal folds where energy production happens. Measurements of oxygen use and ATP output confirmed that the power stations were underperforming, while levels of harmful reactive oxygen species shot up. In fish lacking REP-1, the same pattern emerged: poor glucose uptake, disturbed mitochondrial structure, oxidative stress, and gradual breakdown of the light-sensing outer segments.
A hormone that restores balance
Because the energy crisis began with failed glucose entry, the researchers tested whether leptin, a hormone known to encourage glucose transporter movement in nerve-like cells, could help. In REP-1–deficient retinal cells, leptin treatment pushed GLUT1 and GLUT4 back to the cell surface, revived the insulin-related signaling pathway, and boosted glucose uptake. This, in turn, reduced lipid buildup, improved mitochondrial shape, restored key respiratory proteins, and raised ATP production, even though the cells still lacked REP-1. In the medaka fish model, leptin shifted the lipid profile closer to normal, lowered oxidative stress, improved mitochondrial staining in the eye, and preserved the length and number of cone photoreceptor segments.
What this could mean for future eye treatments
The work suggests that choroideremia is not only a problem of missing gene function, but also of chronic fuel mismanagement inside retinal cells. REP-1 loss pushes cells away from safe sugar use toward risky fat burning, which over time damages their power supply and structure. By showing that a drug like leptin can partially restore glucose use and mitochondrial health in cells and fish, the study highlights metabolic rewiring as a potential treatment route. For people with choroideremia and possibly other retinal dystrophies, therapies that correct this sugar–fat imbalance may one day complement gene-based approaches and help slow the path to blindness.
Citation: Buonocore, S., Giamundo, G., Barone, C. et al. REP-1 deficiency induces aberrant mitochondrial metabolic rewiring from glycolysis to lipid oxidation in CHM disease. Cell Death Dis 17, 436 (2026). https://doi.org/10.1038/s41419-026-08592-6
Keywords: choroideremia, retinal metabolism, mitochondria, glucose uptake, leptin treatment