Clear Sky Science · de
REP-1-Mangel induziert abnorme mitochondriale Stoffwechselumstellung von Glykolyse auf Lipidoxidation bei CHM-Erkrankung
Warum diese Augenforschung wichtig ist
Choroideremie ist eine seltene erbliche Augenerkrankung, die Betroffenen langsam das Sehvermögen raubt, häufig beginnend mit Nachtblindheit in der Kindheit und fortschreitend bis zum vollständigen Sehverlust. Diese Studie zeigt, wie ein einziges fehlendes Protein in Netzhautzellen ihr Energiegleichgewicht durcheinanderbringt und sie dazu zwingt, Fette statt Zucker zu verbrennen. Dieser Wechsel schädigt still und leise die Zellen, die unser scharfes Sehen erhalten, und könnte auf einen neuen Behandlungsansatz für diese und verwandte Augenerkrankungen hinweisen.
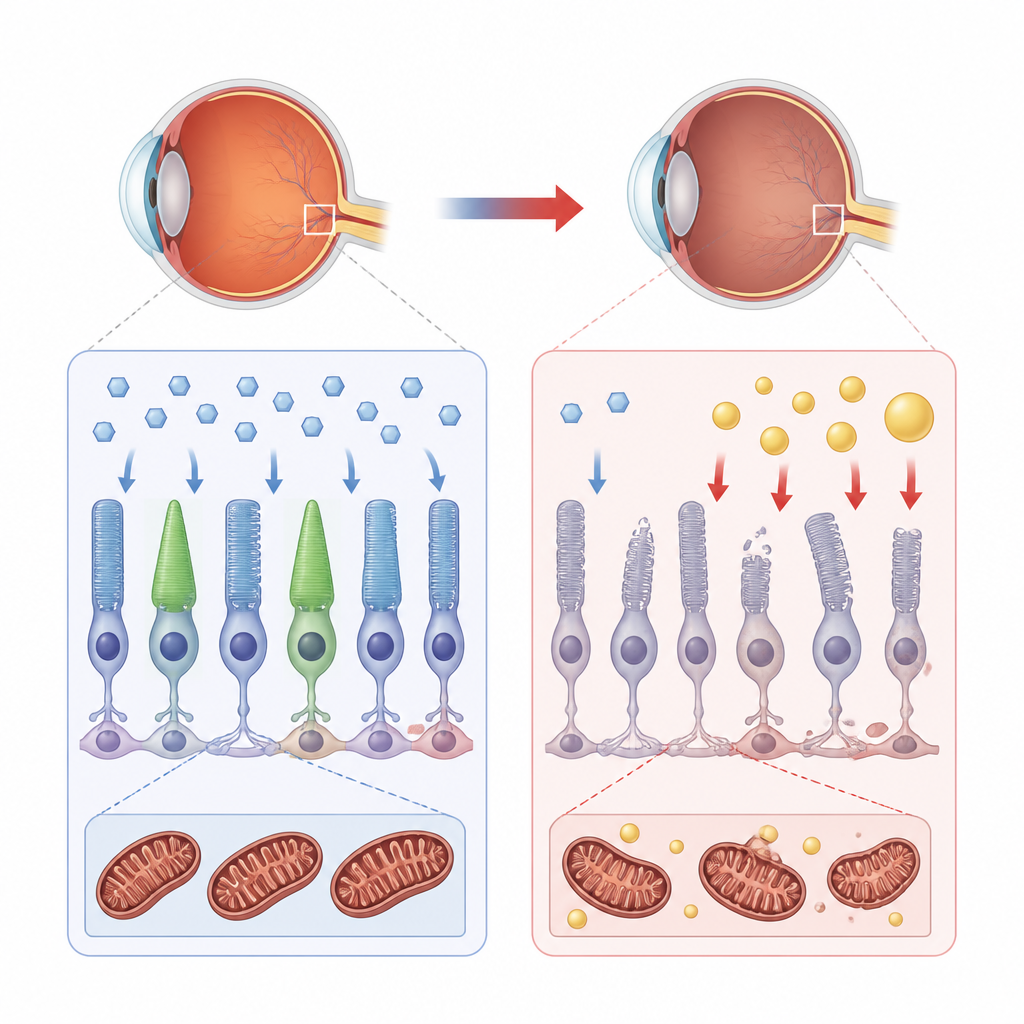
Wie gesunde Netzhautzellen sich mit Energie versorgen
Die Netzhaut ist eines der energiehungrigsten Gewebe des Körpers. Unter normalen Bedingungen stützen sich ihre Stütz- und lichtempfindlichen Zellen hauptsächlich auf Glukose, einen Einfachzucker, der über spezielle Türöffnerproteine, sogenannte Transporter, in die Zelle gelangt. Einmal innen, wird Glukose durch die Glykolyse abgebaut und anschließend in die Mitochondrien eingespeist, die winzigen Kraftwerke, die ATP produzieren, die Energieeinheit der Zelle. Fette können als Ersatzbrennstoff dienen, spielen in einer gesunden Netzhaut aber eher eine unterstützende als eine dominante Rolle.
Der fehlende Helfer, der die Zuckernutzung entgleisen lässt
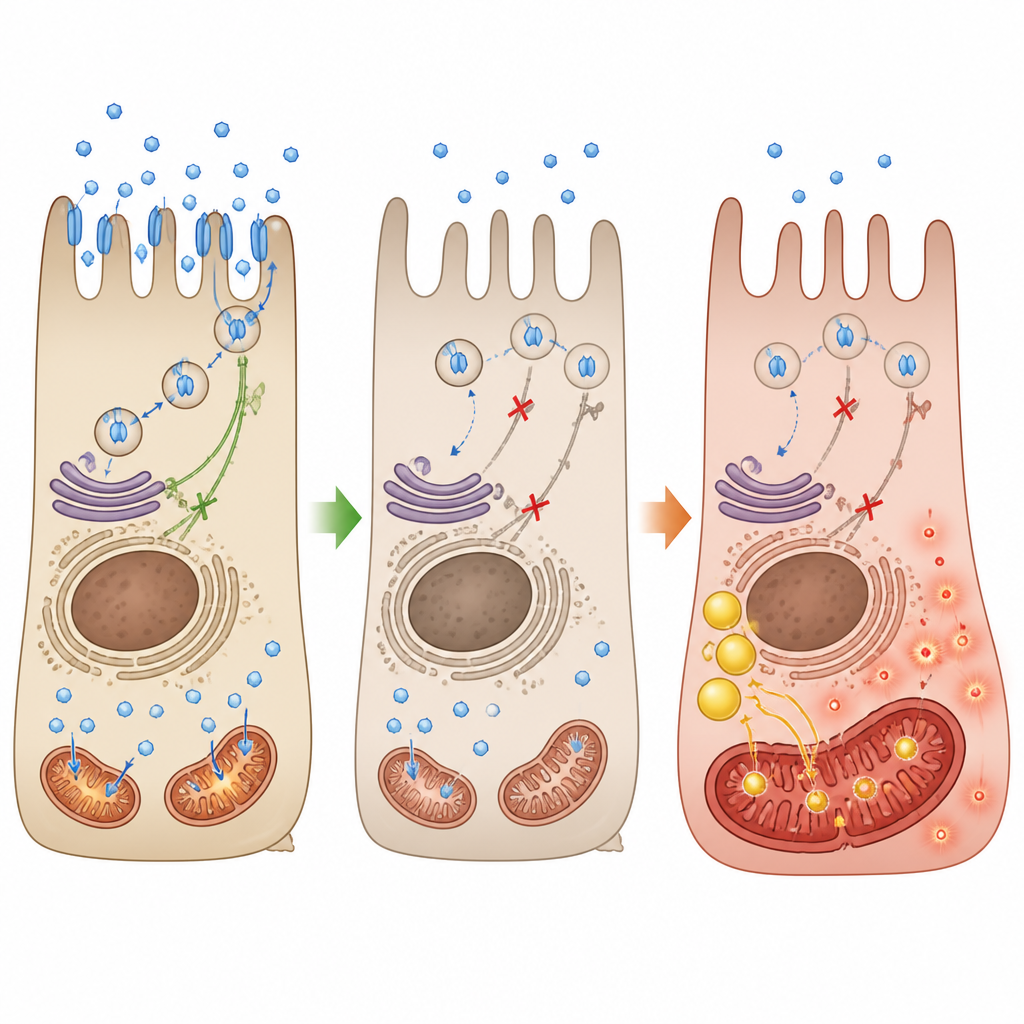
Bei Choroideremie führen Mutationen im CHM-Gen zum Fehlen eines Proteins namens REP-1, das normalerweise beim Intrazellulärtransport vieler Frachtstücke hilft. Die Autoren nutzten menschliche retinal-pigmentierte Zellen und ein kleines Fischmodell, um zu untersuchen, was beim Verlust von REP-1 geschieht. Sie fanden heraus, dass wichtige Glukosetransporter, GLUT1 und GLUT4, entweder in geringerer Menge vorhanden sind oder die Zelloberfläche nicht erreichen. Folglich sinkt die Glukoseaufnahme und insulinähnliche Signale, die normalerweise den Transporter-Transfer zur Membran fördern würden, werden abgeschwächt. Ohne ausreichenden Zuckereintritt stockt der bevorzugte Energiefluss der Zelle.
Wenn die Kraftwerke von Zucker auf Fett umschalten
Ist die Glykolyse beeinträchtigt, verlagern die Netzhautzellen ihre Energiegewinnung hin zur Lipidverbrennung. Das Team beobachtete eine Zunahme von Genen, die mit dem Lipidstoffwechsel verknüpft sind, die Ansammlung von Lipidtröpfchen in den Zellen und höhere Spiegel oxidierter Fette. Mitochondrien verkleinerten sich, verloren ihr normales verzweigtes Netzwerk und zeigten Schäden an den inneren Falten, in denen die Energieproduktion stattfindet. Messungen des Sauerstoffverbrauchs und der ATP-Produktion bestätigten eine verringerte Leistungsfähigkeit der Kraftwerke, während die schädlichen reaktiven Sauerstoffspezies zunahmen. Im REP-1-defizienten Fisch zeigten sich dieselben Muster: schlechte Glukoseaufnahme, gestörte Mitochondrienstruktur, oxidativer Stress und ein schrittweiser Abbau der lichtempfindlichen Außensegmente.
Ein Hormon, das das Gleichgewicht wiederherstellt
Da die Energiekrise mit dem ausgefallenen Glukoseeinstrom begann, testeten die Forscher, ob Leptin — ein Hormon, das bekannt dafür ist, in nervenähnlichen Zellen die Bewegung von Glukosetransportern zu fördern — helfen könnte. In REP-1-defizienten Netzhautzellen sorgte Leptin dafür, dass GLUT1 und GLUT4 wieder an die Zelloberfläche zurückkehrten, der insulinbezogene Signalweg reaktiviert wurde und die Glukoseaufnahme anstieg. Dadurch nahm die Lipidansammlung ab, die mitochondriale Morphologie besserte sich, wichtige Atemproteine wurden wiederhergestellt und die ATP-Produktion stieg, obwohl die Zellen weiterhin REP-1 fehlte. Im Medaka-Fischmodell verschob Leptin das Lipidprofil in Richtung Normalität, verringerte den oxidativen Stress, verbesserte die mitochondriale Markierung im Auge und erhielt Länge und Anzahl der konischen Photorezeptor‑Segmente.
Was das für künftige Augenbehandlungen bedeuten könnte
Die Arbeit legt nahe, dass Choroideremie nicht nur ein Problem verlorener Genfunktion ist, sondern auch eine chronische Fehlsteuerung der Treibstoffnutzung in Netzhautzellen. REP-1-Mangel veranlasst Zellen, sich von der sicheren Zuckernutzung zugunsten riskanter Fettverbrennung zu entfernen, was im Laufe der Zeit Versorgung und Struktur schädigt. Indem gezeigt wird, dass ein Wirkstoff wie Leptin die Glukosenutzung und die mitochondriale Gesundheit in Zellen und Fischen teilweise wiederherstellen kann, rückt die Studie die metabolische Umprogrammierung als potenziellen Behandlungsweg in den Blick. Für Menschen mit Choroideremie und möglicherweise anderen retinalen Dystrophien könnten Therapien, die dieses Zucker‑Fett‑Ungleichgewicht korrigieren, eines Tages Gentherapien ergänzen und helfen, den Weg zur Erblindung zu verlangsamen.
Zitation: Buonocore, S., Giamundo, G., Barone, C. et al. REP-1 deficiency induces aberrant mitochondrial metabolic rewiring from glycolysis to lipid oxidation in CHM disease. Cell Death Dis 17, 436 (2026). https://doi.org/10.1038/s41419-026-08592-6
Schlüsselwörter: Choroideremie, retinaler Stoffwechsel, Mitochondrien, Glukoseaufnahme, Leptinbehandlung