Clear Sky Science · de
Lisinopril aktiviert BI1, um den Lipidstoffwechsel umzuprogrammieren und die Autophagie bei ALS wiederherzustellen
Warum diese Forschung für Familien und Patientinnen und Patienten wichtig ist
Amyotrophe Lateralsklerose (ALS) raubt Menschen die Muskelkraft und letztlich die Fähigkeit zu atmen, während die verfügbaren Therapien nur begrenzte Wirkung zeigen. Diese Studie untersucht, ob ein seit Langem eingesetztes Blutdruckmittel, Lisinopril, zweckentfremdet werden kann, um Nerven- und Muskelzellen bei ALS zu schützen, indem es beeinflusst, wie Zellen mit Fetten umgehen und beschädigte Teile recyceln. Die Arbeit legt nahe, dass ein bekanntes Medikament helfen könnte, die Energiefabriken des Körpers zu stabilisieren, Vernarbung im Muskel zu verringern und krankheitsähnliche Veränderungen in einem etablierten ALS-Mausmodell zu verlangsamen.
Blick auf den Friedensstifter einer Zelle
Die Forschenden konzentrierten sich auf ein Protein namens Bax Inhibitor 1, kurz BI1, das in einer Zellkompartiment sitzt, das Stresssignale steuert. BI1 wurde mit dem Schutz von Hirnzellen in Verbindung gebracht, seine Rolle bei ALS und im Stoffwechsel war jedoch unklar. Durch den Vergleich der Genaktivität in Muskeln gesunder Mäuse, ALS-Mäusen und ALS-Mäusen mit zusätzlichem BI1 fand das Team heraus, dass BI1 viele Wege beeinflusst, die mit Fett- und Zuckerstoffwechsel, Zellüberleben und Gewebsvernarbung zusammenhängen. BI1 dämpfte ein starkes Signalsystem, das durch das Molekül TGF-beta angetrieben wird und Entzündung sowie Fibrose fördert, und hob zugleich Blockaden des zellulären Aufräumprozesses Autophagie auf.
Wie ein Blutdruckmittel ins Spiel kam
Um ein Medikament zu finden, das BI1 ohne Gentherapie stärken kann, nutzte das Team Computerwerkzeuge, um die Struktur des BI1-Proteins zu modellieren und eine Bibliothek bereits für den Menschen zugelassener Wirkstoffe zu durchsuchen. Lisinopril zeigte sich als starker Bindungspartner in einer spezifischen Tasche an BI1. In nervenähnlichen Zellen mit einer ALS-assoziierten SOD1-Mutation erhöhte Lisinopril die BI1-Spiegel, half, die elektrische Stabilität der Mitochondrien zu erhalten, und reduzierte Anzeichen programmierter Zellsterblichkeit. Es bewahrte außerdem das innere Gerüst und die Länge der Nervenfortsätze, was darauf hindeutet, dass behandelte Zellen besser in der Lage sein könnten, ihre Verbindungen zu Muskeln aufrechtzuerhalten.
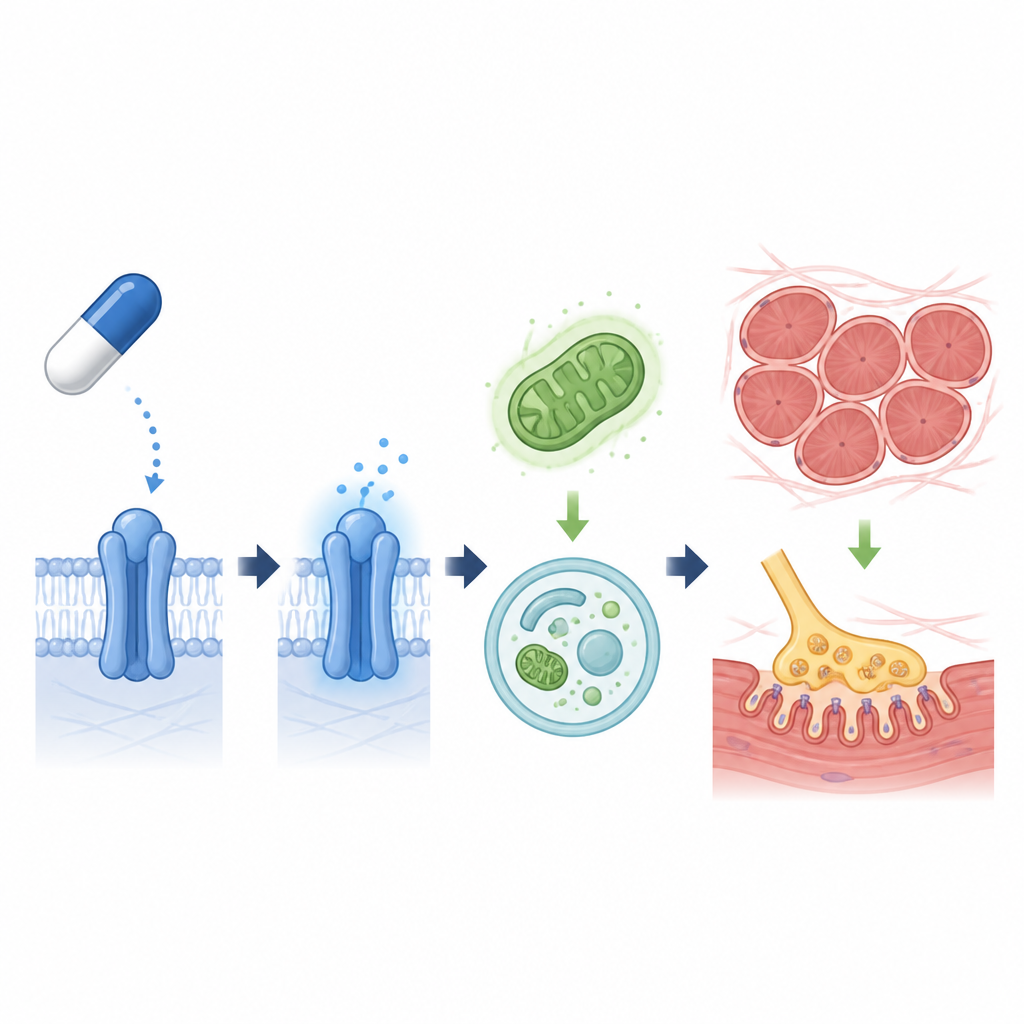
Reparatur des zellulären Aufräums und Verringerung von Gewebevernarbung
Innerhalb der Zellen müssen beschädigte Proteine und verschlissene Strukturen abgebaut und recycelt werden. Bei ALS ist dieses Aufräumsystem häufig träge. Die Studie zeigte, dass Lisinopril über BI1 TGF-beta und verwandte Signale senkte, die normalerweise die Autophagie abschalten. Mit dem Nachlassen dieser Bremsen stiegen Marker aktiver Autophagie, und ein wichtiger Wachstumsweg, PI3K/AKT/mTOR, wurde gedämpft. In Mausmuskelgewebe verschob Lisinopril außerdem die Genaktivität weg von der Bildung starrer Narben und verringerte die Ansammlung von Typ-I-Kollagen und Fibrosemarkern. Gleichzeitig enthielten behandelte Muskeln mehr langsam zuckende, ausdauerorientierte Fasern, mehr Mitochondrien und höhere Speicher an zellulärem Brennstoff, was auf eine gesündere Energiebalance hinweist.
Neuausrichtung von Fetten und Schutz der Nerven in ALS-Mäusen
Lipidomik, eine großangelegte Untersuchung von Fettmolekülen, zeigte, dass Lisinopril die Fettlandschaft in Muskeln von ALS-Mäusen umgestaltete. Das Medikament verschob das Gleichgewicht weg von Membranlipiden wie bestimmten Sphingolipiden und Glycerophospholipiden, die Mitochondrien schädigen und Entzündungen fördern können. Es erhöhte bestimmte Speicherfette, darunter Triglyceride mit butyratähnlichen Fragmenten, die mit besserer Mitochondrienfunktion in Verbindung stehen, und verringerte Lipide, die mit oxidativem Stress und Zelltod assoziiert sind. In lebenden ALS-Mäusen verzögerte Lisinopril den Krankheitsbeginn, verlängerte die Lebenszeit, unterstützte Laufleistung und Griffstärke, verringerte Muskelabbau und bewahrte die Struktur von Rückenmarksneuronen, deren isolierender Myelinschicht und den neuromuskulären Verbindungsstellen, an denen Nerven mit Muskeln kommunizieren.
Was das für die zukünftige ALS-Behandlung bedeuten könnte
Insgesamt deuten die Ergebnisse darauf hin, dass Lisinopril BI1 in Muskeln und Nervenzellen aktivieren, die TGF-beta-Signalisierung dämpfen, das zelluläre Aufräumen wiederherstellen und die Lipidnutzung in ein Muster lenken kann, das die Energieversorgung unterstützt statt Zellschäden zu fördern. In einem ALS-Mausmodell führte dieses vielschichtige Wirkungsbild zu langsamerer Degeneration von Nerven und Muskeln und zu besserer körperlicher Leistungsfähigkeit. Obwohl weitere Untersuchungen in größeren Tieren und beim Menschen nötig sind, skizziert die Studie eine detaillierte Karte, wie ein bekanntes Herzmedikament neu genutzt werden könnte, um ALS über die Beeinflussung von Stoffwechsel und Zellresilienz zu adressieren, statt nur einzelne Krankheitswege anzusprechen.
Zitation: Yin, H., Ren, Z., Zhang, Y. et al. Lisinopril activates BI1 to reprogram lipid metabolism and restore autophagy in ALS. Commun Biol 9, 705 (2026). https://doi.org/10.1038/s42003-026-09930-2
Schlüsselwörter: amyotrophe Lateralsklerose, lisinopril, Lipidstoffwechsel, Autophagie, Neuroprotektion