Clear Sky Science · de
Systematische Analyse von snRNA-Genen zeigt häufige RNU2-2-Varianten bei dominanten und rezessiven Entwicklungs- und epileptischen Enzephalopathien
Warum winzige RNA-Stücke für die Gehirnentwicklung von Kindern wichtig sind
Ärztinnen und Ärzte können heute nahezu jeden Buchstaben der DNA eines Kindes lesen, trotzdem verlassen viele Kinder mit schwerer Entwicklungsverzögerung und Epilepsie die Klinik ohne klare Diagnose. Diese Studie richtet das Augenmerk auf einen überraschend kleinen und zuvor vernachlässigten Teil unseres genetischen Codes: winzige RNA-Gene, die Zellen dabei helfen, Botschaften zu schneiden und zusammenzusetzen, bevor diese in Proteine übersetzt werden. Die Autorinnen und Autoren zeigen, dass Veränderungen in einem solchen Gen, genannt RNU2-2, eine häufige Ursache schwerer neuroentwicklicher Störungen sind und bis zu etwa 1 von 300 betroffenen Kindern treffen können.
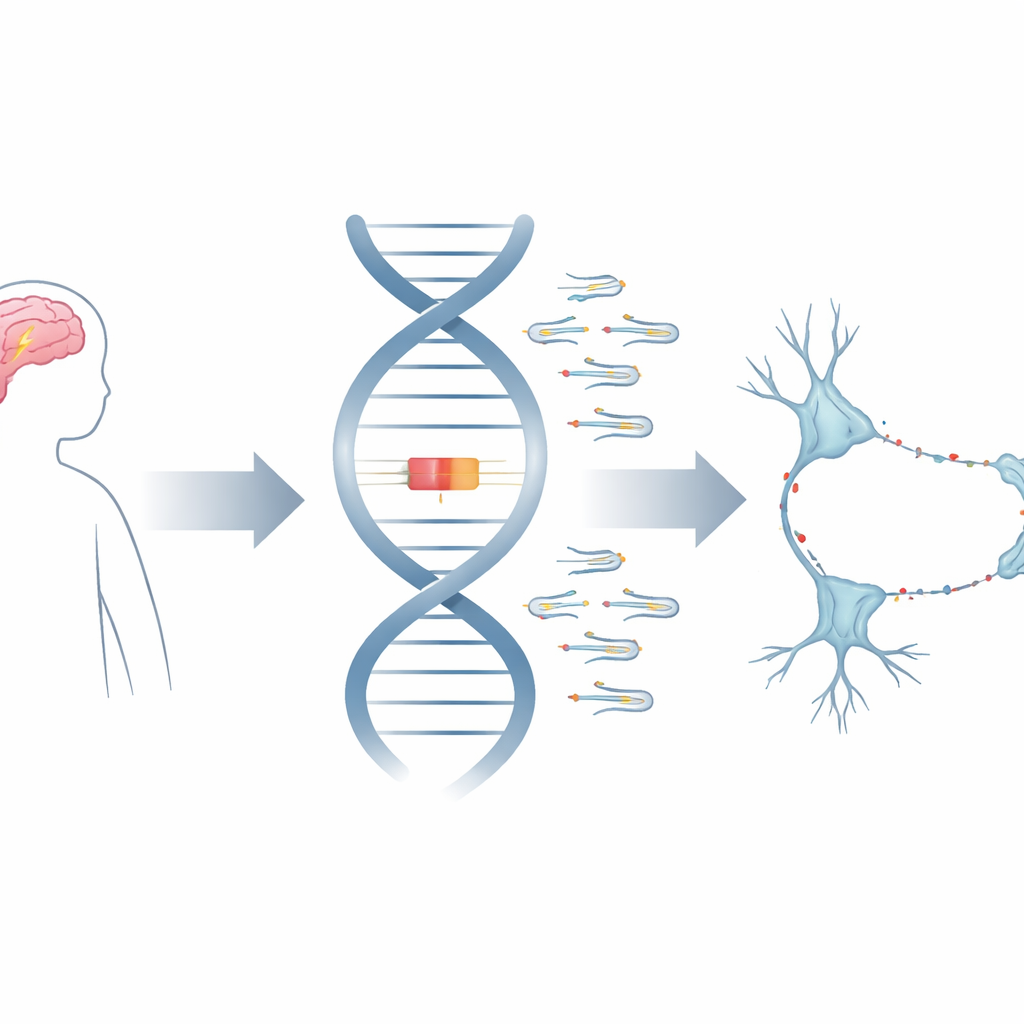
Eine verborgene Ebene des genetischen Handbuchs
Die meisten Gentests konzentrieren sich auf proteinkodierende Gene, doch unsere Zellen sind auch auf viele kurze RNA-Moleküle angewiesen, die niemals Proteine werden. Dazu gehören die kleinen nukleären RNAs, oder snRNAs, die den Kern des Spliceosoms bilden — der molekularen Maschine, die rohe RNA-Botschaften zurechtschneidet und die nützlichen Teile zusammenfügt. Das Gen RNU2-2 produziert eine Variante der U2-snRNA, ein zentrales Element, das hilft, den exakten Schnittpunkt in der RNA zu bestimmen. Da viele Kopien von snRNA-Genen existieren und sehr ähnlich aussehen, waren sie schwer zu untersuchen und wurden häufig als inaktive „Pseudogene“ abgetan.
Durchforsten von Tausenden Genomen nach aktiven snRNA-Genen
Das Team durchforstete zunächst über 2.000 annotierte snRNA-Gene im menschlichen Genom und nutzte öffentlich zugängliche Gehirn-RNA-Daten sowie regulatorische Karten, um wahrscheinliche funktionelle Gene von inaktiven Relikten zu trennen. Dieser Aufwand identifizierte 200 snRNA-Gene, die funktionell erscheinen, viele davon zuvor als Pseudogene eingestuft. Anschließend durchsuchten die Forschenden Genomdaten von mehr als 34.000 Menschen mit seltenen Erkrankungen in Frankreich und konzentrierten sich dabei auf ungewöhnliche neue Mutationen, die nur beim Kind auftreten, sowie auf Mutationspaare, die von beiden Elternteilen vererbt wurden. Auffällig hoben sich Varianten in RNU2-2 in beiden Analysen ab, insbesondere bei Personen mit neuroentwicklichen Störungen.
Eine häufige Ursache für Entwicklungsverzögerung und Epilepsie
Durch das Zusammenführen von Daten aus Frankreich und internationalen Partnerzentren versammelten die Autorinnen und Autoren 141 Patientinnen und Patienten aus 122 Familien mit Veränderungen in RNU2-2. Fünfunddreißig Kinder trugen eine von zwei wiederkehrenden Mutationen, die dominant wirken — das heißt, eine einzelne veränderte Kopie des Gens reicht aus, um die Erkrankung zu verursachen. Eine noch größere Gruppe — 91 betroffene Individuen aus 73 Familien — trug schädigende Varianten auf beiden Kopien des Gens, was eine rezessive Form der Erkrankung enthüllt, die mindestens doppelt so häufig ist wie die dominante Form. Unabhängig von der Vererbung wiesen nahezu alle Betroffenen Entwicklungsverzögerung und geistige Beeinträchtigung auf, und etwa 85 % entwickelten Epilepsie, meist beginnend vor dem Alter von drei Jahren. Viele zeigten außerdem autistische Merkmale, Bewegungsstörungen und subtile, aber wiederkehrende Gesichtsmerkmale.
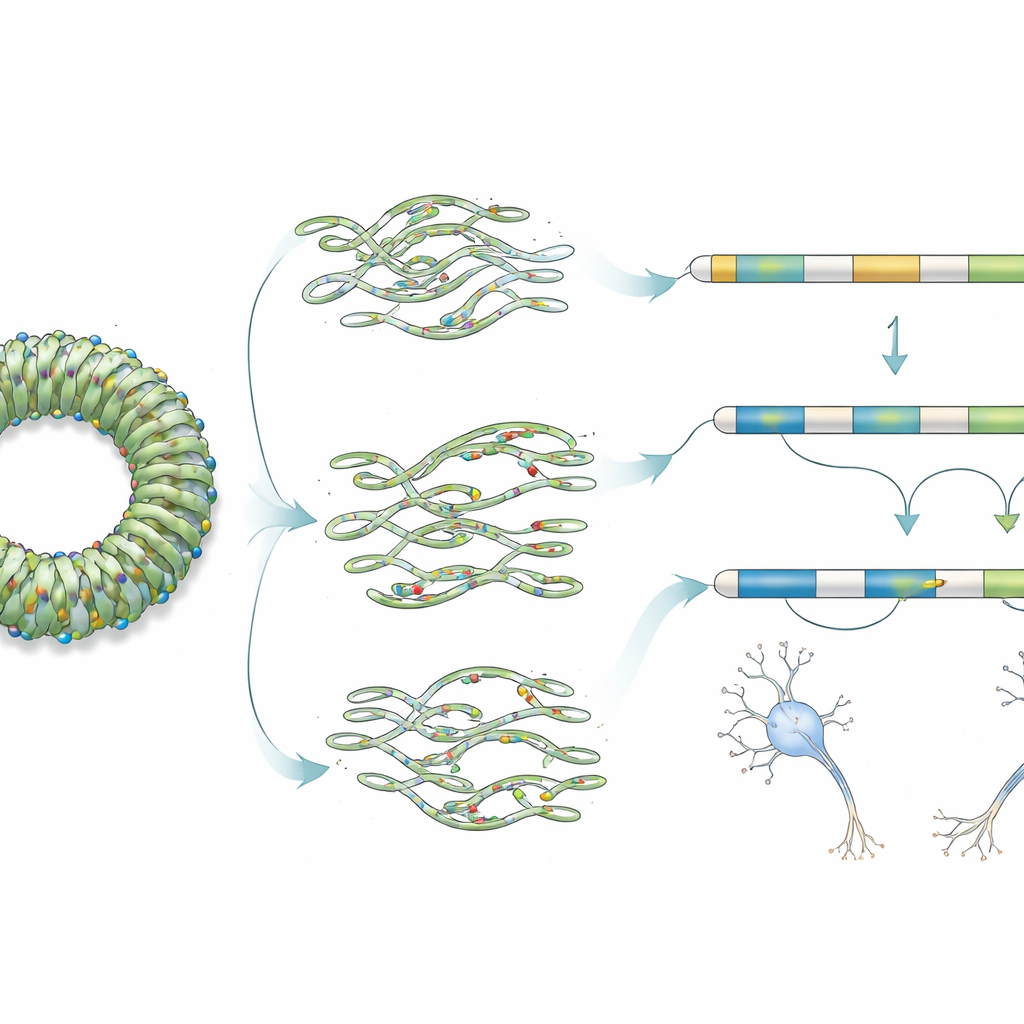
Wie kleine RNA-Veränderungen die Gehirnfunktion stören
Um zu verstehen, wie RNU2-2-Varianten Zellen schaden, kartierten die Forschenden jede Mutation auf detaillierten dreidimensionalen Modellen der U2-RNA im Spliceosom. Einige Veränderungen liegen in Regionen, die der U2 helfen, den korrekten „Branchpunkt“ in der RNA zu erkennen, andere treffen strukturelle Elemente, die zum Aufbau und Import der RNA in den Zellkern nötig sind. Die Autorinnen und Autoren zeigen, dass bestimmte Varianten die U2-Funktion wahrscheinlich vollständig zerstören, während andere sie teilweise abschwächen. Bei der Untersuchung von Blutzellen der Patientinnen und Patienten fanden sie nur subtile Veränderungen: einige Exons wurden etwas häufiger übersprungen, und DNA-Methylierungsmuster — ein chemisches Markierungssystem auf der DNA — zeigten leicht veränderte, variantenspezifische Muster. Diese moderaten Signaturen passen zur Idee, dass RNU2-2-Mutationen die RNA-Verarbeitung über viele Gene hinweg nur leicht stören, mit besonders starken Folgen im sich entwickelnden Gehirn, wo RNU2-2 stärker aktiv ist.
Ein Kontinuum zwischen dominanter und rezessiver Erkrankung
Eine der interessantesten Beobachtungen ist, dass sich dominante und rezessive Formen der RNU2-2-Erkrankung klinisch bemerkenswert ähnlich zeigen. Anstatt zwei völlig getrennte Krankheiten schlagen die Autorinnen und Autoren ein Modell des „Wirkungsgradienten“ vor. Sehr zerstörerische Varianten an zentralen funktionellen Stellen können die Erkrankung allein verursachen und dominant wirken. Mildere Veränderungen haben möglicherweise kaum Auswirkungen, es sei denn, das Kind erbt eine zweite schädigende Variante auf der anderen Genkopie, was eine rezessive Erkrankung hervorruft. Wiederum können andere Kombinationen — etwa eine starke und eine schwächere Variante — den Schweregrad der Symptome modulieren. Da snRNA-Gene wie RNU2-2 eine ungewöhnlich hohe Rate neuer Mutationen akkumulieren, treten diese unterschiedlichen genetischen Szenarien in der Population häufig auf.
Was das für Familien und zukünftige Forschung bedeutet
Für Familien, die nach Antworten suchen, zeigt diese Arbeit, dass RNU2-2-Varianten zusammen etwa 0,35 % aller Neuroentwicklungsstörungen ausmachen — wodurch dieses winzige RNA-Gen zu einem der am häufigsten beteiligten nichtkodierenden Gene bei kindlichen Hirnerkrankungen wird, vergleichbar in seiner Bedeutung mit dem kürzlich entdeckten ReNU-Syndrom-Gen RNU4-2. Die Studie ermahnt Kliniker zudem, bei Entdeckung einer neuen Mutation in RNU2-2 oder verwandten snRNA-Genen sorgfältig nach einer zweiten verborgenen Variante zu suchen, anstatt automatisch von einem rein dominanten Effekt auszugehen. Allgemeiner deuten die Ergebnisse darauf hin, dass viele andere „nichtkodierende“ RNA-Gene unerkannte Ursachen unerklärter Entwicklungsstörungen sein könnten. Mit verbesserten Genomsequenzierungs- und Long-Read-Technologien könnte die systematische Untersuchung dieser bislang übersehenen Regionen eine neue Klasse häufiger, bisher unsichtbarer genetischer Ursachen schwerer kindlicher Epilepsie und geistiger Behinderung offenbaren.
Zitation: Leitão, E., Santini, A., Cogne, B. et al. Systematic analysis of snRNA genes reveals frequent RNU2-2 variants in dominant and recessive developmental and epileptic encephalopathies. Nat Genet 58, 782–797 (2026). https://doi.org/10.1038/s41588-026-02547-5
Schlüsselwörter: neuroentwicklungsstörungen, epileptische Enzephalopathie, Spliceosom, nichtkodierende RNA, RNU2-2-Varianten