Clear Sky Science · de
Fehlreguliertes nukleares Lamin B1 bei DYT1-Dystonie verdickt die Kernlamina und stört 14-3-3-Proteine
Wenn das innere Skelett einer Zelle versagt
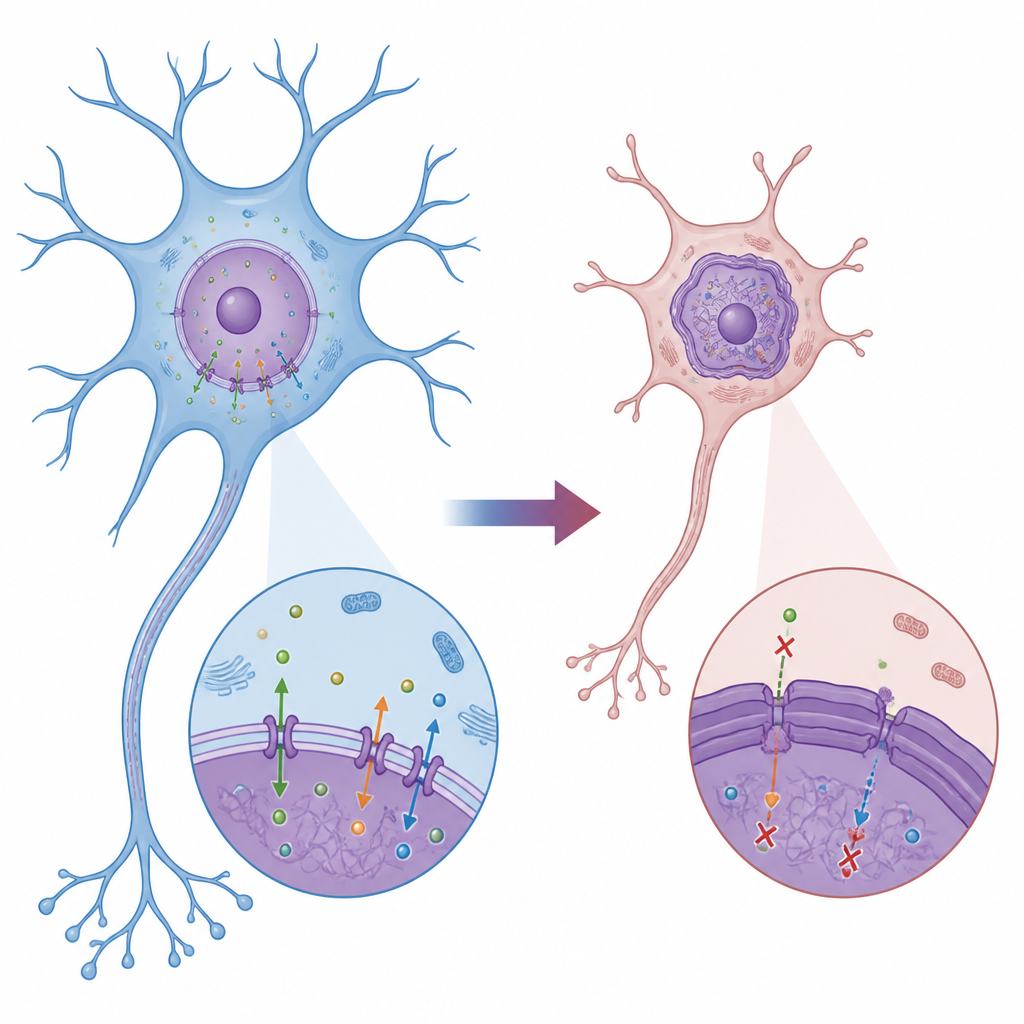
Die in der Kindheit beginnende DYT1-Dystonie ist eine seltene Bewegungsstörung, die Muskeln verdrehen und die Körperhaltung verkrümmen kann. Ihre Ursache liegt jedoch tief in den Gehirnzellen, an der Grenze des Zellkerns, der unsere DNA enthält. Diese Studie zeigt, wie ein Strukturprotein namens Lamin B1, Teil der „Kernschale“ der Zelle, bei DYT1-Dystonie fehlreguliert wird, den Kern verformt, den molekularen Verkehr blockiert und dabei Hilfsproteine stört, die Nervenzellen für Wachstum und Funktion benötigen.
Ein genauerer Blick auf eine kindliche Bewegungsstörung
DYT1-Dystonie beginnt meist in der Kindheit oder Jugend, einer wichtigen Phase für die Hirnschaltkreise, die Bewegung steuern. Die meisten Fälle beruhen auf einer winzigen dreibasenpaarigen Deletion im TOR1A-Gen, das TorsinA codiert, ein Enzym, das Struktur und Transportsysteme rund um den Kern unterstützt. Frühere Arbeiten an menschlichen Neuronen von Patientinnen und Patienten deuteten darauf hin, dass Lamin B1, ein zentraler Bestandteil der nukleären Lamina an der Innenseite der Kernmembran, überreichlich vorhanden und fehlplatziert ist. Die aktuelle Studie zielte darauf ab, genau zu verstehen, wie dieses Lamin-B1-Problem die Kernform und -funktion verändert und wie diese Veränderungen sich auf die Entwicklung der Neuronen auswirken.
Verformte Kerne und verstopfter molekularer Verkehr
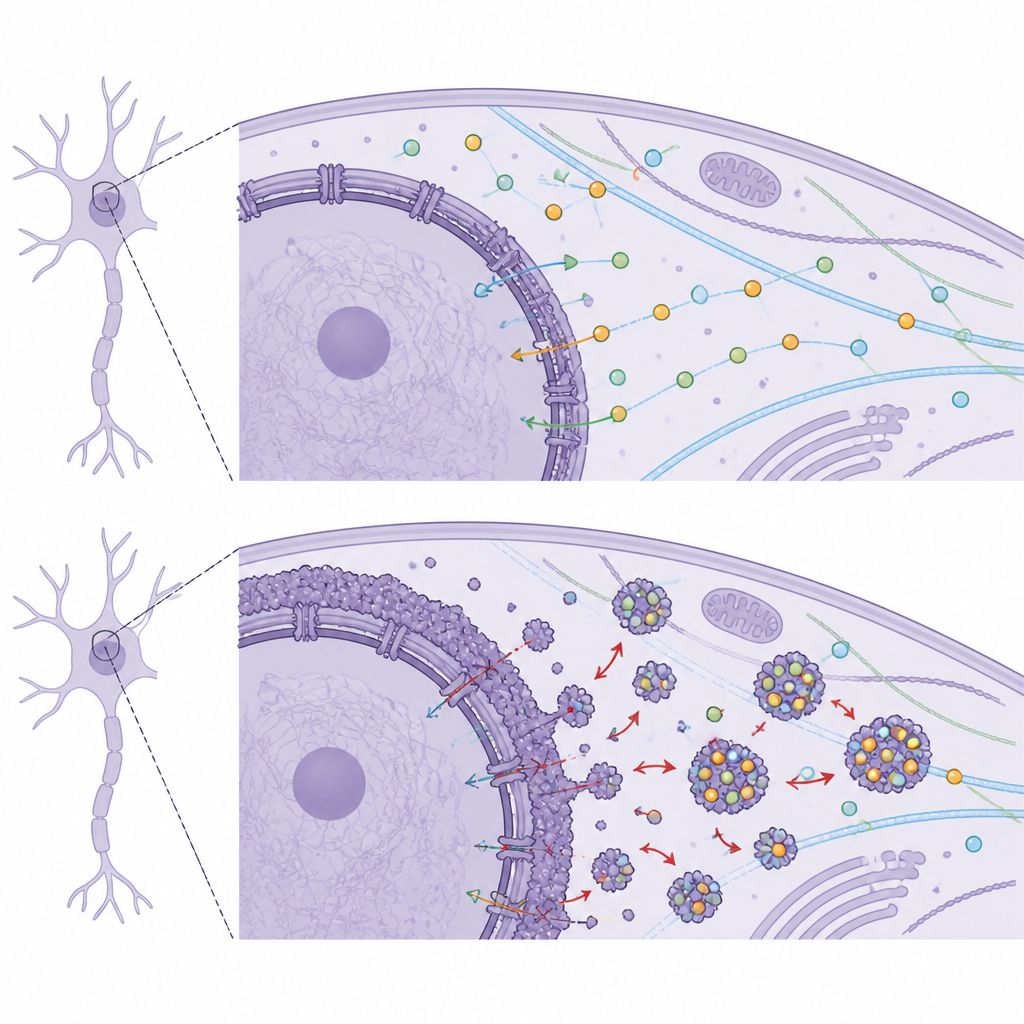
An Hand von Fibroblasten von DYT1-Patienten und altersangepassten gesunden Kontrollen fanden die Forschenden, dass Zellen mit der DYT1-Mutation größere, missgestaltete Kerne und ungewöhnlich starke Lamin-B1-Färbung aufwiesen, während ein verwandtes Protein, Lamin A/C, normal erschien. Elektronenmikroskopie zeigte einen verdickten dunklen Streifen direkt unter der inneren Kernmembran, was darauf hindeutet, dass die Kernlamina selbst ungewöhnlich massiv und starr geworden war. Biochemische Tests bestätigten, dass ein Teil des Lamin B1, das normalerweise am Kernrand verankert bleibt, in das umgebende Zytoplasma ausgelaufen war. Diese Veränderungen standen im Zusammenhang mit schweren Transportproblemen: Fluoreszenzmarkierungen und RNA-Sonden zeigten, dass sowohl Proteine als auch Boten-RNAs Schwierigkeiten hatten, die Kerngrenze zu passieren, sich auf der falschen Seite anreicherten und damit die übliche Informationszirkulation zwischen Kern und Zytoplasma gestört war.
Wie fehlplatziertes Lamin B1 die Neuronenmaschinerie stört
Um zu untersuchen, was fehlplatziertes Lamin B1 „festhalten“ könnte, nutzte das Team humane, aus Stammzellen gewonnene Motoneurone und ein auf Neuroblastom basierendes Neuronenmodell, brachte Lamin B1 ins Zytoplasma und zog dann seine Bindungspartner heraus. Massenspektrometrie identifizierte Hunderte von interagierenden Proteinen, die an RNA-Verarbeitung, Proteinsynthese, Energiestoffwechsel, Zytoskelettorganisation und insbesondere am Nukleocytoplasmatischen Transport beteiligt sind. Viele dieser Partner sind für neuronenspezifische Aufgaben wie Axonwachstum, Synapsenbildung und Signalübertragung entscheidend. Auffällig war eine Gruppe: die 14-3-3-Proteine, eine Familie häufiger Chaperone, die andere Proteine während der Gehirnentwicklung führen und stabilisieren und deren zelluläre Lokalisation regulieren. Zytoplasmatisches Lamin B1 band mehrere 14-3-3-Varianten deutlich stärker als normal, was darauf hindeutet, dass es diese Helfer von ihren richtigen Aufgaben wegsequestriert.
Hilfsproteine, die wachsende Neurone formen
Die Forschenden fragten weiter, was passiert, wenn 14-3-3-Proteine selbst hoch- oder runterreguliert werden. In gesunden, aus Stammzellen abgeleiteten Motoneuronen führte die Reduktion zweier wichtiger 14-3-3-Varianten (bekannt als beta und gamma) zu kürzeren Neuriten und weniger Verzweigungen sowie zu geringeren Spiegeln von Genen, die für die neuronale Reifung wichtig sind. In DYT1-Neuronen waren mehrere 14-3-3-Gene natürlicherweise reduziert, und Lamin B1 war sichtbar in Neuriten fehlgelagert. Als das Team 14-3-3 beta oder gamma überexprimierte, wuchsen DYT1-Neuronen mit längeren, stärker verästelten Prozessen und zeigten höhere Werte von Reifungsmarkern. Gleichzeitig verschob sich Lamin B1 zurück in Richtung Kern, seine zytoplasmatische Anhäufung nahm ab, und sowohl Protein- als auch mRNA-Transport über die Kerngrenze verbesserten sich, insbesondere der Import von Proteinen in den Kern.
Was das für das Verständnis und die Behandlung von Dystonie bedeutet
Einfach ausgedrückt verbindet diese Arbeit ein fehlerhaftes nukleäres „Gerüst“ mit gestörtem Neuronenwachstum bei DYT1-Dystonie. Zu viel Lamin B1 und Lamin B1 am falschen Ort verdickt und versteift die Kernschale, verformt den Kern, verstopft den molekularen Verkehr und bindet 14-3-3-Hilfsproteine, die Neurone für ihre Entwicklung benötigen. Durch die Erhöhung der 14-3-3-Spiegel konnten die Forschenden diesen Zustand teilweise entwirren: Lamin B1 nahm wieder eine normalere Position ein, der nukleäre Transport verbesserte sich und das Neuritenwachstum wurde gefördert. Obwohl diese Ergebnisse aus Zellmodellen und nicht direkt aus Patientinnen und Patienten stammen, weisen sie Lamin B1 und 14-3-3-Proteine als vielversprechende Ziele für künftige Therapien aus, die gefährdete Motoneurone bei DYT1-Dystonie und möglicherweise anderen neurologischen Erkrankungen mit beschädigter Kernarchitektur schützen oder wiederherstellen könnten.
Zitation: Duan, Y., Sepehrimanesh, M., Hosain, M.A. et al. Dysregulated nuclear Lamin B1 in DYT1 dystonia thickens nuclear lamina and disrupts 14-3-3 proteins. Cell Death Discov. 12, 245 (2026). https://doi.org/10.1038/s41420-026-03090-2
Schlüsselwörter: DYT1-Dystonie, Lamin B1, nukleärer Transport, 14-3-3-Proteine, Motoneurone