Clear Sky Science · zh
赖氨酸145位点的乳酰化促进KAT8–TIP60复合物形成,从而促进p53在赖氨酸120位点的乙酰化及其促凋亡功能
这项研究为何重要
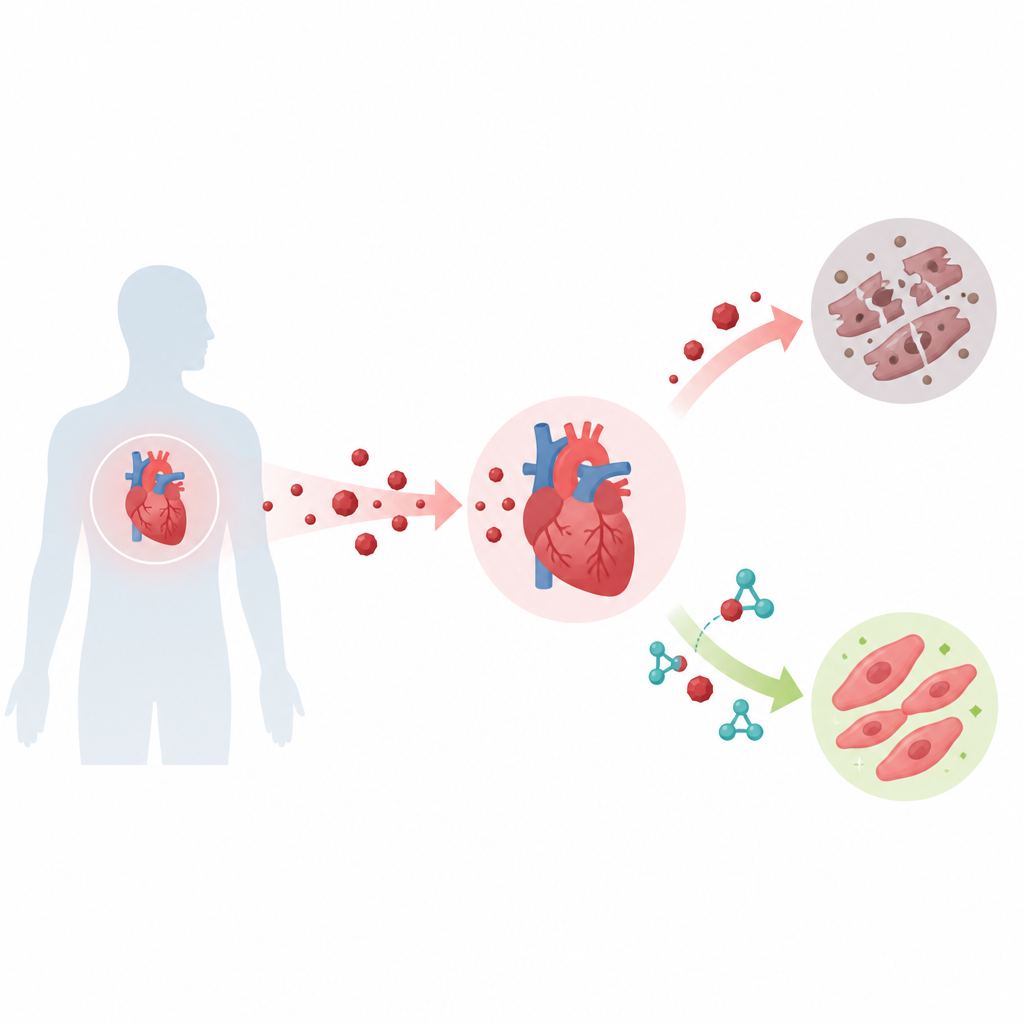
抗癌药能挽救生命,但有时会牺牲心脏健康。该研究解开了心肌细胞内一条隐秘的化学连锁反应,这条链部分解释了为何阿霉素会损害心脏,并指出一种已有的降糖药格列美脲,可能在不削弱其抗癌效力的情况下保护心脏。
细胞内的分子开关
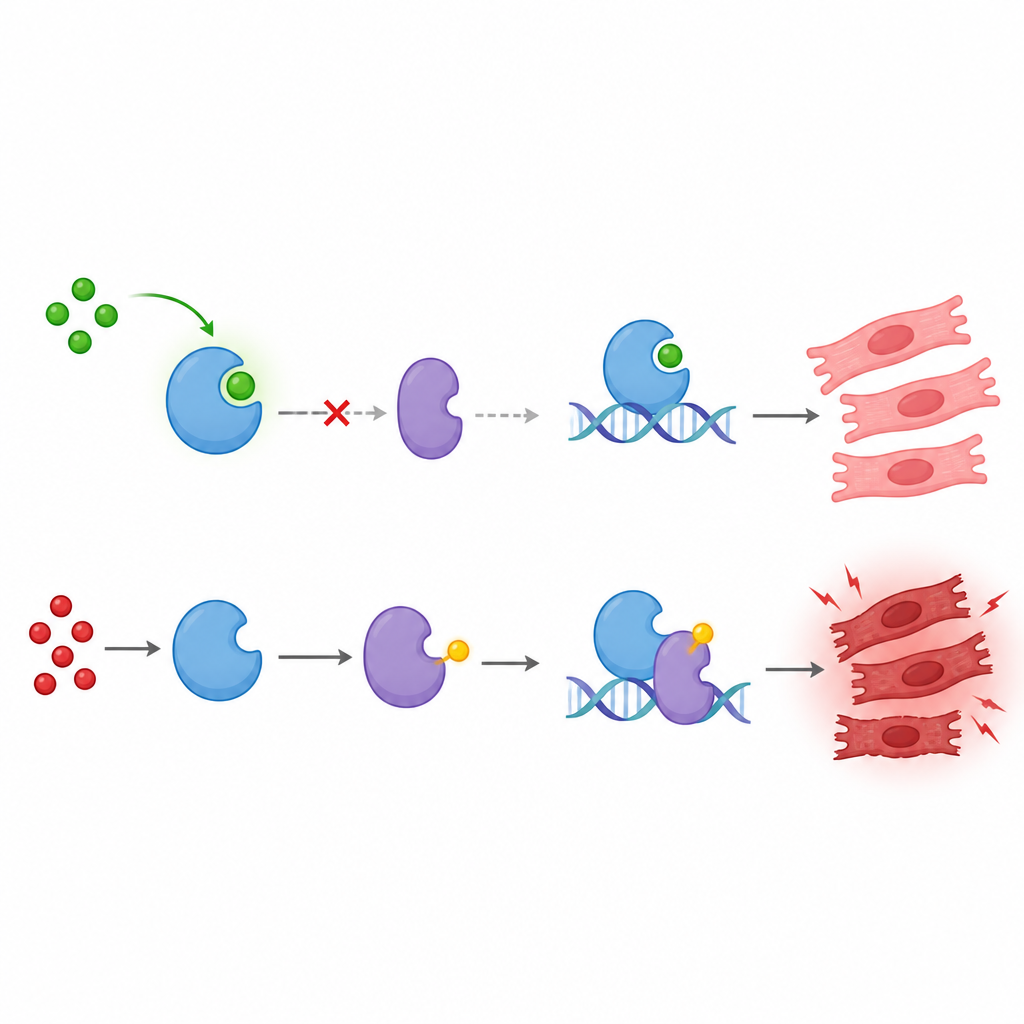
我们的细胞通过微小的化学标记不断调整蛋白的功能,这些标记像开关一样控制活性。研究团队关注一种较新的标记——乳酰化,它与细胞将糖分解为乳酸的代谢过程有关。他们研究了调控多种细胞过程的蛋白KAT8,探讨它是否被乳酰化以及乳酰化的意义。结果发现KAT8主要在单一位点被乳酰化——赖氨酸145,而这种修饰依赖于细胞内乳酸水平。提升葡萄糖浓度会增加乳酸并促进KAT8乳酰化,阻断葡萄糖代谢则减少该修饰,表明这一标记直接响应细胞代谢状态。
将代谢与细胞“安全官”连接起来
接着,研究者将这一代谢开关与p53联系起来。p53常被称为细胞的“守护者”,负责判断受损细胞是修复还是自我毁灭。他们发现当KAT8在赖氨酸145位被乳酰化时,更强烈地促进p53在赖氨酸120位的乙酰化。该乙酰化标记使p53更擅长激活驱动细胞自杀的基因。研究还鉴定出GCN5和SIRT6分别为KAT8乳酰化的“写入”和“擦除”酶:GCN5添加乳酰基,SIRT6去除它。调节这些酶的活性会改变KAT8的乳酰化,从而影响p53的乙酰化。
触发细胞死亡的特殊蛋白团队
这种效应的关键并非来自KAT8自身的酶学活性,而是取决于它选择与谁组队。赖氨酸145处的乳酰化有助于KAT8与另一蛋白TIP60结合。KAT8与TIP60形成复合物,与p53结合并高效地使p53在赖氨酸120位乙酰化。被改造的p53随后更强烈地结合调控两个强效促死基因BAX和PUMA的DNA开关,增强它们的表达。无法在赖氨酸145处被乳酰化的KAT8表现出较弱的KAT8–TIP60结合、较少的p53乙酰化、p53在BAX和PUMA上的结合减少,以及这些促死蛋白的表达下降。
从分子通路到心脏损伤
阿霉素是一种广泛使用的抗癌药物,但会引起持续的心脏损伤。在人源心肌样细胞和小鼠模型中,作者证明阿霉素显著增加KAT8在赖氨酸145位的乳酰化、增强KAT8–TIP60的互作、提高p53在赖氨酸120位的乙酰化并升高BAX和PUMA水平,所有这些改变共同推动心肌细胞死亡。当他们阻断KAT8在该位点的乳酰化时,阿霉素诱导的细胞死亡明显减少,p53活性及其靶基因的异常升高也被抑制。在小鼠中,阿霉素处理增加了心脏损伤的指标、导致心脏变小并提升心肌组织的细胞死亡,均与该通路在体内发挥作用相一致。
将一种糖尿病药再利用以保护心脏
为了找到实际可行的中断这一有害链条的方法,研究组在药物数据库中筛选能够结合KAT8乳酰化位点附近的小分子,发现已有用于2型糖尿病治疗的格列美脲能强烈结合KAT8。格列美脲与GCN5竞争结合KAT8,削弱二者相互作用并阻止KAT8在赖氨酸145处的乳酰化。继而减少了KAT8–TIP60复合物的形成、p53在赖氨酸120位的乙酰化以及BAX和PUMA的激活。在心肌细胞和小鼠中,格列美脲降低了阿霉素诱导的心肌细胞死亡和损伤指标,保护了心脏大小,并且在乳腺癌模型中并未降低阿霉素抑制肿瘤生长的能力。
对患者意味着什么
通俗地说,该研究揭示了心肌细胞内一条被隐藏的接力:糖代谢通过乳酰基标记使KAT8上锁,帮助其招募TIP60,从而增强p53的促死活性。阿霉素劫持了这条接力,推动心肌细胞走向自毁。通过阻断KAT8上的乳酰化标记,格列美脲有效削弱了p53对促死基因的驱动并减轻了心脏损伤。尽管仍需更多工作,包括基因学模型和临床试验,但这些发现为在不牺牲抗癌疗效的前提下保护心脏提供了一条有希望的途径。
引用: Liu, H., Li, Z., Lei, D. et al. Lactylation at lysine 145 fosters KAT8-TIP60 complex formation to promote p53 acetylation at lysine 120 and its pro-apoptotic function. Nat Commun 17, 4442 (2026). https://doi.org/10.1038/s41467-026-71108-5
关键词: p53, 乳酰化, 阿霉素心脏毒性, KAT8, 格列美脲