Clear Sky Science · en
Lactylation at lysine 145 fosters KAT8-TIP60 complex formation to promote p53 acetylation at lysine 120 and its pro-apoptotic function
Why this study matters
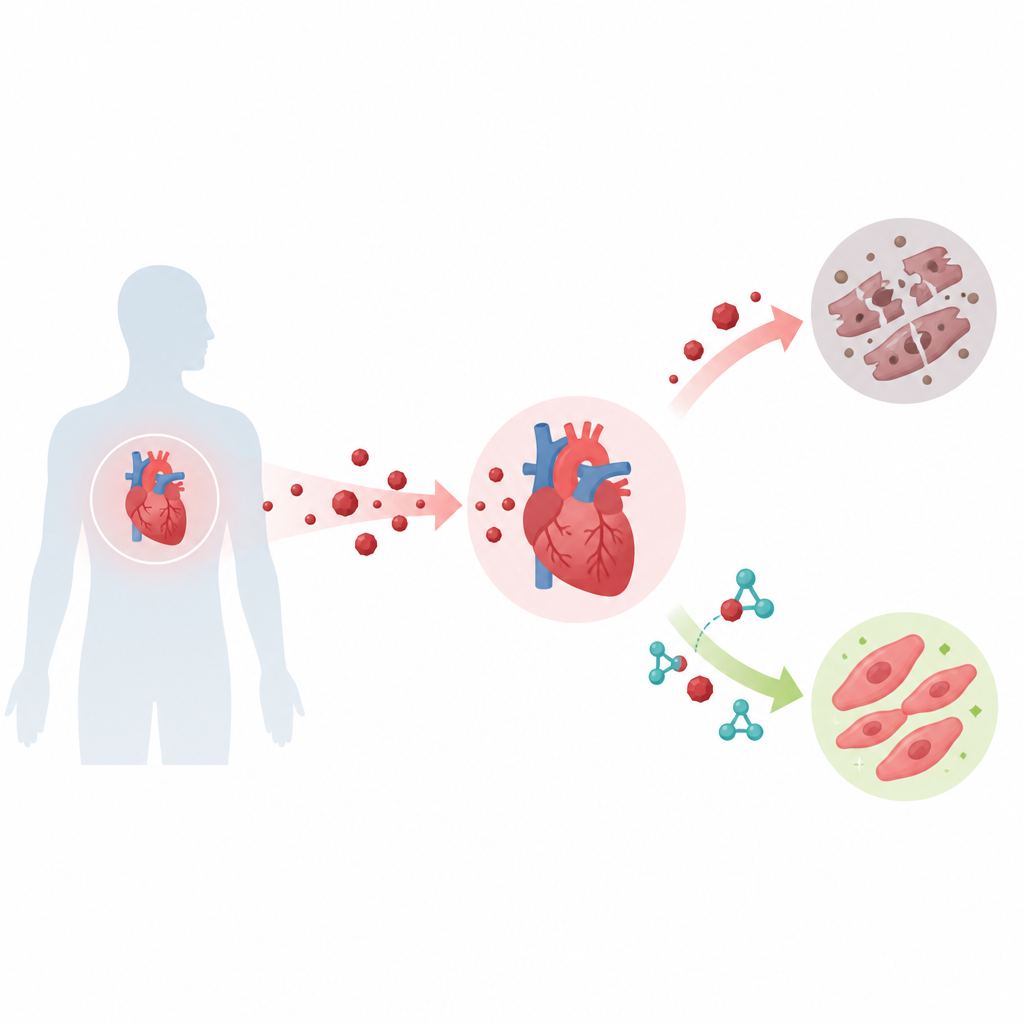
Cancer drugs can save lives but sometimes leave the heart worse off. This study unravels a hidden chemical chain reaction inside heart cells that helps explain why the drug doxorubicin can damage the heart, and it points to an existing diabetes medicine, glimepiride, as a possible way to shield the heart without dulling the cancer-fighting effect.
A molecular switch inside our cells
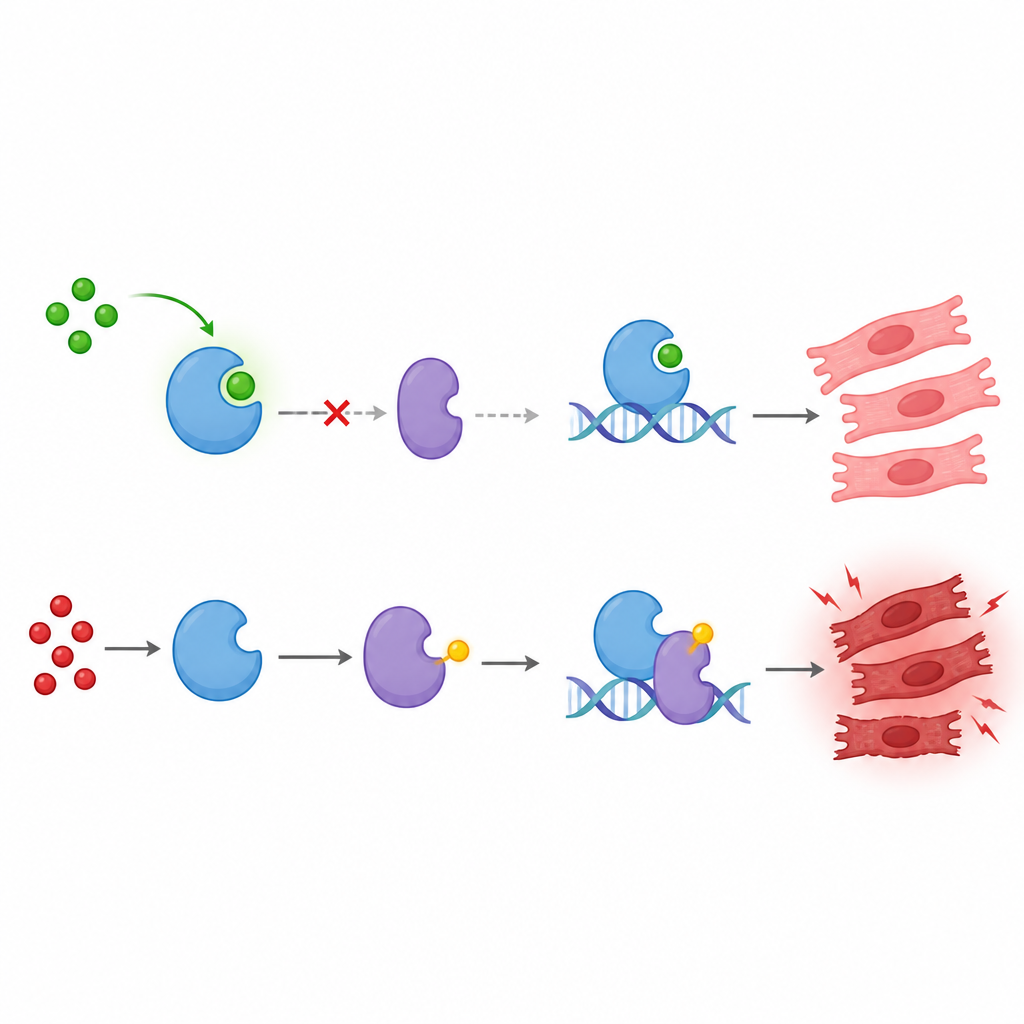
Our cells constantly tweak the behavior of their proteins with tiny chemical tags that act like on and off switches. The team focused on a newer kind of tag called lactylation, which is linked to the cell’s breakdown of sugar into lactate. They studied a protein called KAT8, known for controlling many cell processes, and asked whether it is lactylated and what that means. They discovered that KAT8 is mainly lactylated at a single spot, an amino acid called lysine 145, and that this modification depends on the cell’s lactate levels. Raising glucose increased lactate and boosted KAT8 lactylation, while blocking glucose metabolism reduced it, showing that this tag responds directly to cellular metabolism.
Connecting metabolism to the cell’s safety officer
Next the researchers connected this metabolic switch to p53, a protein often called the cell’s “guardian” because it helps decide whether a damaged cell should repair itself or die. They found that when KAT8 is lactylated at lysine 145, it more strongly promotes the addition of another chemical tag, an acetyl group, to p53 at lysine 120. This particular acetyl mark makes p53 better at turning on genes that drive cell suicide. The enzymes GCN5 and SIRT6 were identified as writers and erasers of the KAT8 lactyl mark: GCN5 adds the lactyl group, while SIRT6 removes it. Changing the activity of these enzymes tuned KAT8 lactylation and, in turn, p53 acetylation.
A special protein team that triggers cell death
The key to this effect turned out not to be KAT8’s own enzyme activity but whom it chooses to team up with. Lactylation at lysine 145 helps KAT8 bind another protein, TIP60. Together, KAT8 and TIP60 form a complex that associates with p53 and efficiently acetylates p53 at lysine 120. This upgraded form of p53 then binds more strongly to the DNA switches that control two potent pro-death genes, BAX and PUMA, boosting their activity. Cells in which KAT8 cannot be lactylated at lysine 145 show weaker KAT8–TIP60 binding, less p53 acetylation, poorer docking of p53 on BAX and PUMA, and lower levels of these death-promoting proteins.
From molecular pathway to heart damage
Doxorubicin is a widely used cancer drug, but it can cause lasting heart injury. In human heart-like cells and in mice, the authors showed that doxorubicin sharply increases KAT8 lactylation at lysine 145, strengthens the KAT8–TIP60 partnership, raises p53 acetylation at lysine 120, and heightens BAX and PUMA levels, all of which drive heart cell death. When they prevented KAT8 from being lactylated at this site, doxorubicin triggered far less cell death, and the dangerous rise in p53 activity and its target genes was blunted. In mice, doxorubicin treatment increased markers of heart injury, caused smaller hearts, and boosted cell death in heart tissue, all consistent with this pathway operating in living animals.
Repurposing a diabetes drug for heart protection
Seeking a practical way to interrupt this harmful chain, the team searched a drug database for molecules that could bind near KAT8’s lactylation site. They identified glimepiride, a drug already used to treat type 2 diabetes, as a strong binder of KAT8. Glimepiride competes with GCN5 for access to KAT8, weakening their interaction and preventing KAT8 lactylation at lysine 145. This in turn reduces KAT8–TIP60 complex formation, p53 acetylation at lysine 120, and BAX and PUMA activation. In heart cells and in mice, glimepiride lowered doxorubicin-induced heart cell death and injury markers, preserved heart size, and did not reduce the drug’s ability to slow tumor growth in a breast cancer model.
What this means for patients
In everyday terms, the study reveals a hidden relay inside heart cells in which sugar metabolism primes KAT8 with a lactyl tag, helping it recruit TIP60 to supercharge p53’s pro-death activity. Doxorubicin hijacks this relay, pushing heart cells toward self-destruction. By blocking the lactyl tag on KAT8, glimepiride effectively loosens p53’s grip on cell-death genes and softens the heart damage. While more work, including genetic models and clinical testing, is needed, these findings highlight a promising route to make powerful cancer treatments safer for the heart.
Citation: Liu, H., Li, Z., Lei, D. et al. Lactylation at lysine 145 fosters KAT8-TIP60 complex formation to promote p53 acetylation at lysine 120 and its pro-apoptotic function. Nat Commun 17, 4442 (2026). https://doi.org/10.1038/s41467-026-71108-5
Keywords: p53, lactylation, doxorubicin cardiotoxicity, KAT8, glimepiride