Clear Sky Science · zh
QSOX2 的非酶学功能直接调控 JUNB–ITGB4 轴并增强 EGFR 突变型肺腺癌对奥希替尼的耐药性
为何某些肺癌对一种关键药物失去反应
奥希替尼是一种已延长许多常见类型肺癌患者寿命的口服药物,但几乎所有肿瘤最终都会攻克它。本研究探讨某些肺癌细胞如何重编程其内部信号,使其即便在药物存在下仍能继续生长,并指出未来治疗可能利用的新薄弱环节。
隐藏在肺肿瘤内的帮手
研究者聚焦于肺腺癌——最常见的肺癌类型,尤其是那些肿瘤带有 EGFR 基因改变的患者。奥希替尼针对这些异常的 EGFR 分子,但肿瘤常通过备份通路获得耐药。通过分析患者样本和肿瘤的单细胞数据,团队发现一种鲜为人知的蛋白 QSOX2 在癌细胞中异常高表达,而在邻近的正常肺组织中并不明显。QSOX2 水平较高与疾病更具侵袭性以及第三代 EGFR 药物(如奥希替尼)获益时间更短相关。
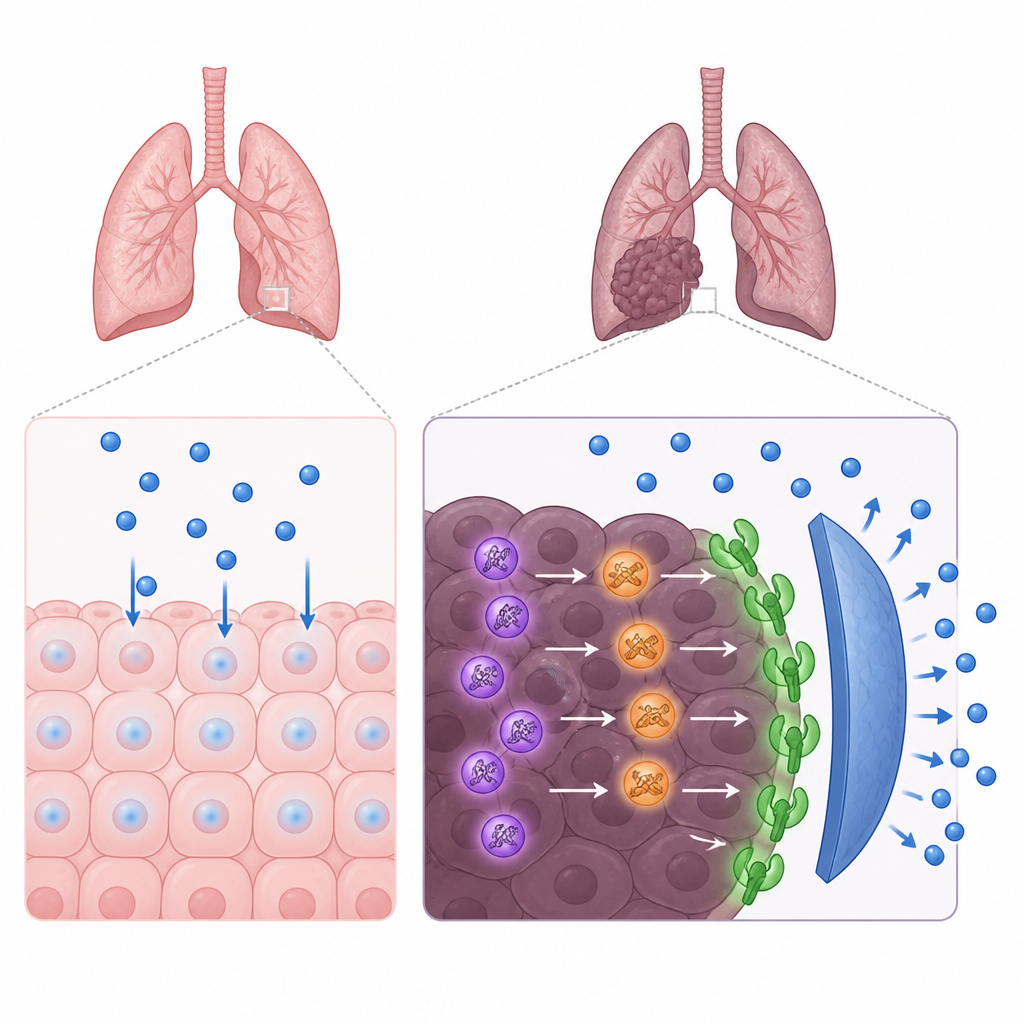
QSOX2 帮助癌细胞摆脱药物抑制
为验证 QSOX2 是否主动促成肿瘤的耐药性,科学家在携带 EGFR 突变的肺癌细胞系中调节其表达。当降低 QSOX2 时,细胞对奥希替尼更敏感;当提高 QSOX2 时,细胞需要超过 20 倍的药物剂量才能被抑制。患者血液检测显示,那些疾病更早进展的患者血清中 QSOX2 水平更高。这些发现合在一起表明,QSOX2 不仅是侵袭性癌症的旁观者标志物,还是耐药的功能性驱动因子。
一条增强存活信号的蛋白链
深入研究时,团队揭示了一条从 QSOX2 开始并在细胞表面导致更强存活信号的事件链。QSOX2 与另一种控制基因活性的蛋白 JUNB 发生物理结合,这种结合稳定了 JUNB 并促使其进入细胞核,在核内 JUNB 激活 ITGB4 基因的表达。膜上的额外 ITGB4 随后启动一对称为 FAK 和 AKT 的信号分子,这些分子是已知的促生长和促存活因子。反过来,AKT 帮助维持核内 JUNB 的活性,形成一个自我增强的循环,即使 EGFR 被奥希替尼抑制,该旁路仍然被持续打开。
在患者来源的迷你肿瘤中证实的耐药性
体外实验有时不能完全反映真实肿瘤,因此研究者在更现实的模型中检验了他们的假设。他们从患者样本培养三维迷你肿瘤(类器官),并将人类肿瘤移植到小鼠体内建立患者来源的异种移植模型。在这两种系统中,对奥希替尼不再响应的肿瘤显示出高水平的 QSOX2、JUNB 和 ITGB4,而敏感肿瘤则水平较低。阻断 FAK 或 AKT,或降低 QSOX2 或 ITGB4 表达,均使耐药类器官和肿瘤对药物更敏感,支持该信号轴在真实世界耐药中的核心作用。
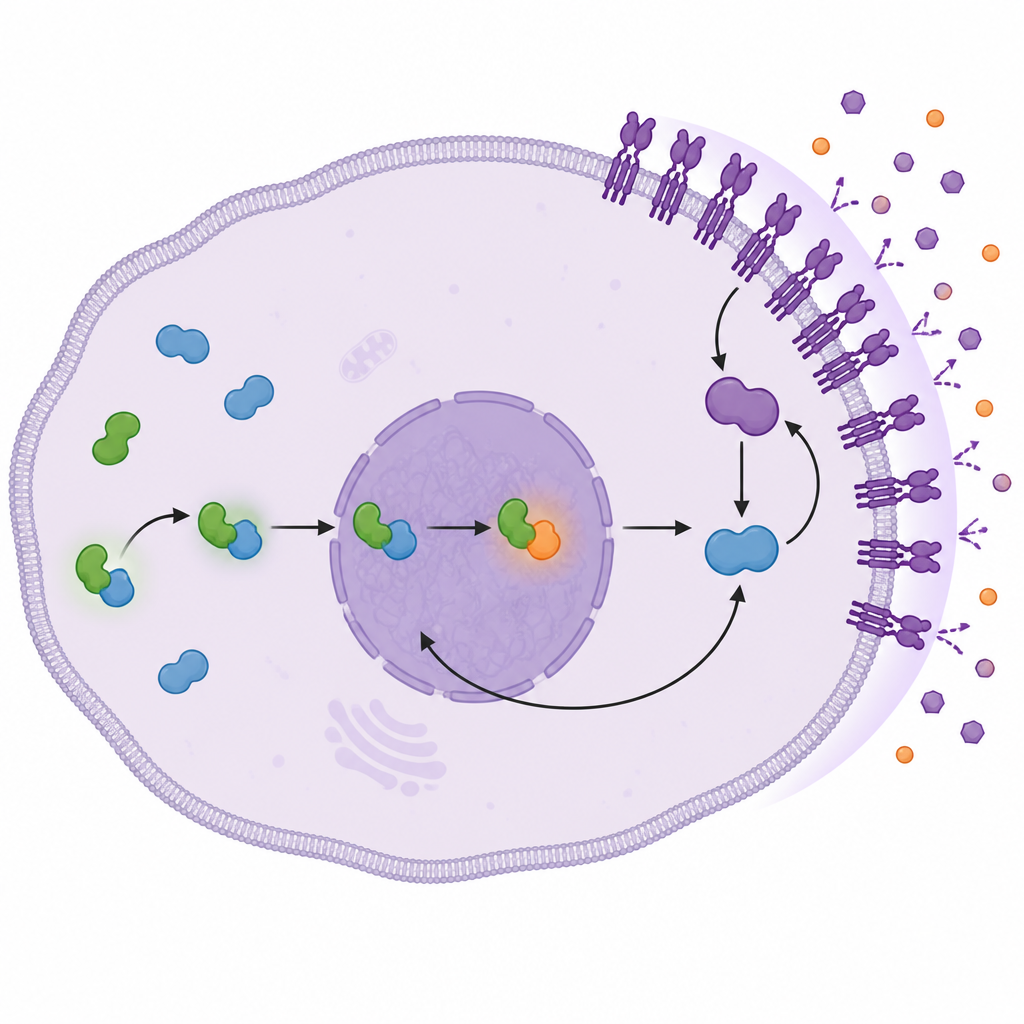
一个可以延长疗效的新靶点
对于 EGFR 突变型肺腺癌患者来说,主要结论是:围绕 QSOX2、JUNB 和 ITGB4 建立的药物旁路环路可驱动对奥希替尼的耐药,通过激活与 EGFR 无关的强大生长信号实现。尽管目前尚无专门阻断 QSOX2 的药物,这项工作提示检测 QSOX2 及相关蛋白可能有助于预测患者从 EGFR 靶向药物中获益的时长,并且未来针对该轴或其下游伙伴 FAK 与 AKT 的治疗可能有助于恢复药物敏感性,延长现有疗法的有效期。
引用: Liu, C., Wang, S., Qi, R. et al. Non-enzymatic function of QSOX2 directly regulates the JUNB-ITGB4 axis and enhanced resistance to osimertinib in EGFR-mutation lung adenocarcinoma. Cell Death Discov. 12, 215 (2026). https://doi.org/10.1038/s41420-026-02969-4
关键词: 奥希替尼 耐药, EGFR 突变型肺癌, QSOX2, ITGB4 FAK AKT 通路, 药物耐受