Clear Sky Science · zh
ASH2L 通过依赖 H3K4me3 的 ITGA6/ERK 信号通路诱导 ER 阳性乳腺癌的他莫昔芬耐药
为何部分乳腺癌停止对关键药物产生反应
许多激素敏感型乳腺癌患者服用他莫昔芬,这种口服药物阻断雌激素的促生长信号。对大多数人来说疗效良好——但有一部分令人担忧的患者,其肿瘤最终会逃避免疫并复发。本研究发现了一个名为 ASH2L 的分子“主谋”,它帮助癌细胞躲避他莫昔芬并表现出更像难以根除的干细胞特性,同时提出了一种可能关闭这种耐药性的药物联合策略。
肿瘤 DNA 中的隐秘开关
ASH2L 是一组负责在 DNA 包装蛋白上添加小型化学标签的蛋白复合体的一员,通过这些修饰微妙地开启或关闭基因。研究者分析了大规模患者数据,发现 ASH2L 基因在雌激素受体(ER)阳性乳腺癌中经常发生扩增——被额外复制。ASH2L 活性异常升高的肿瘤更容易复发并且更致命,即便患者接受了他莫昔芬治疗。有趣的是,ASH2L 的水平并不简单地与雌激素受体水平同步,表明其通过不同途径推动风险。
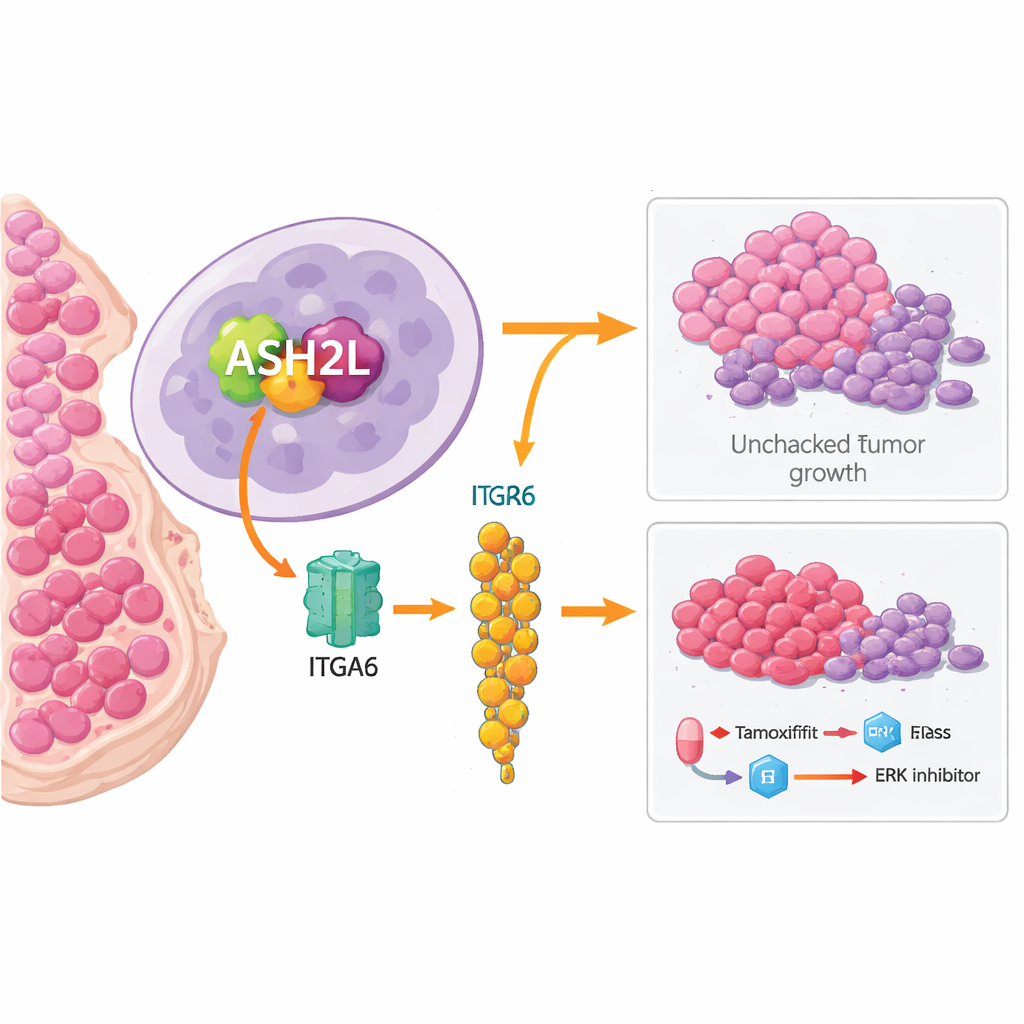
从基因标记者到耐药驱动因子
为了弄清 ASH2L 在细胞内的实际作用,团队在多种 ER 阳性乳腺癌细胞系及小鼠模型中对其进行过表达或抑制。过度表达 ASH2L 会让癌细胞生长更快、形成更多克隆,并在动物体内更容易形成肿瘤。这些 ASH2L 高表达的细胞对他莫昔芬也更难被消灭:它们能耐受更高剂量,治疗后形成更多存活克隆,并呈现更少的程序性细胞死亡信号。当在天然耐药的细胞中降低 ASH2L 时,则出现相反效应——细胞对他莫昔芬更敏感,且小鼠体内的肿瘤在药物作用下缩小更多。
滋养一群顽固的“类干”细胞
研究还发现 ASH2L 能扩增一小部分具有干细胞样特征的癌细胞——这些细胞能再生肿瘤并常常对治疗毫不在意。通过专门的检测,科学家表明提高 ASH2L 会增加具有高酶活性和典型乳腺癌干细胞表面标志的细胞比例,并提升它们在培养中以悬浮球体形式生长的能力。降低 ASH2L 则显著减少这些类干群体,并使注射的细胞更难在小鼠体内启动肿瘤,表明 ASH2L 为一股尤其危险的细胞储备提供燃料。
分子接力:从 ASH2L 到 ITGA6 再到 ERK
在机制研究中,研究者追踪到一条链式反应。ASH2L 附着于两个基因 HIF2A 和 ITGA6 的调控区,并改变附近核小体的化学标记,从而有利于这些基因的激活。HIF2A 又进一步促进 ITGA6 的增强表达。ITGA6 蛋白位于细胞表面并激活一种被称为 ERK 的内部信号通路,ERK 在促进细胞生长和存活方面众所周知。在 ASH2L 高表达的细胞中,ITGA6 与 ERK 活性升高,下游推动细胞周期推进和抑制细胞死亡的蛋白也随之增加。当团队敲低 ITGA6 或用靶向抑制剂阻断 ERK 时,ASH2L 所驱动的类干特性和耐药性在很大程度上消失。
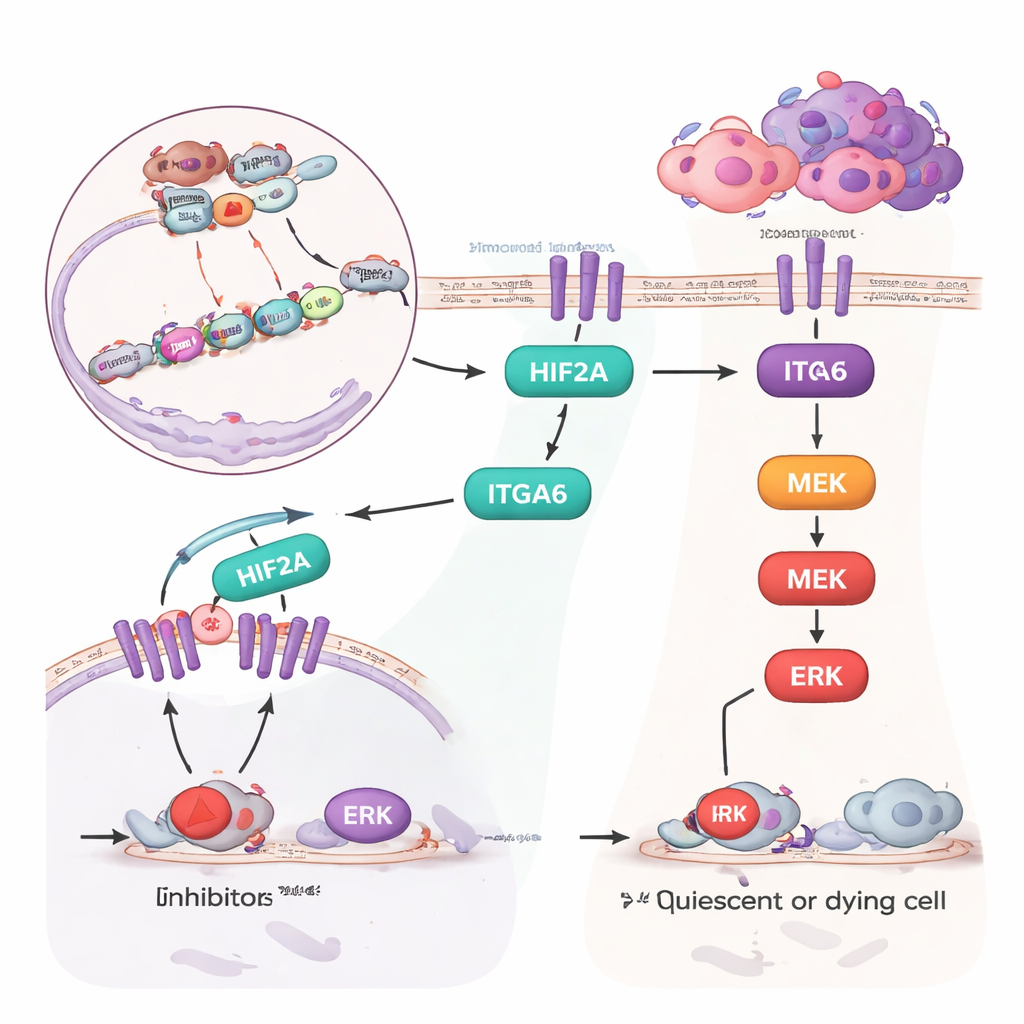
联合用药以破解耐药
既然 ERK 信号被证明是这一路径的关键终点,科学家们便探讨是否将他莫昔芬与 ERK 抑制剂联合使用能克服 ASH2L 驱动的耐药。在细胞培养中,他莫昔芬或 ERK 抑制剂单用对 ASH2L 高表达细胞影响有限,但二者合用显著降低了细胞存活率和克隆形成能力。在携带 ASH2L 过表达肿瘤的小鼠中,他莫昔芬单用几乎无法抑制肿瘤生长,ERK 抑制剂单用也效果甚微。然而两者联合却将肿瘤缩小到与对照肿瘤(未过表达 ASH2L)接受他莫昔芬治疗时相似的程度。
对患者意味着什么
该研究表明 ASH2L 作为一个表观遗传学的主控开关,帮助 ER 阳性乳腺癌对他莫昔芬产生耐药并维持一批类干细胞。通过激活 ITGA6 和 ERK 通路,ASH2L 使肿瘤细胞在激素治疗下仍能持续分裂并避免死亡。结果指向两个有前景的思路:将 ASH2L 水平作为标志物来识别他莫昔芬可能失效的患者,以及使用他莫昔芬与 ERK 抑制剂的联合治疗来重新使耐药肿瘤恢复敏感。尽管仍需更多临床研究,但这项工作勾勒出从 DNA 水平的调控因子到恢复一种沿用数十年的乳腺癌药物功效的可行策略的清晰路径。
引用: Kye, YH., Moon, SJ., Cha, HR. et al. ASH2L induces tamoxifen resistance via H3K4me3 dependent ITGA6/ERK signaling in ER-positive breast cancer. Br J Cancer 134, 1150–1165 (2026). https://doi.org/10.1038/s41416-026-03347-8
关键词: 他莫昔芬耐药, ER 阳性乳腺癌, ASH2L, 癌症干细胞, ERK 信号