Clear Sky Science · sv
En genomisk metod för korrekt identifiering av nära besläktade arter med nästa generations sekvenseringsdata
Varför detta är viktigt för gårdar och bortom
Modern DNA‑sekvensering kan läsa djurs genetiska kod med förbluffande detalj, men även kraftfulla datorer kan få problem med en förvånansvärt grundläggande fråga: kommer dessa sekvenser från ett får eller en get? För lantbrukare, uppfödare, naturvårdare och forskare kan förväxling av arter i stora DNA‑datamängder förstöra studier om hälsa, produktivitet och evolution. Denna artikel presenterar ett enkelt men smart sätt att skilja åt nära besläktade arter—demonstrerat på får och getter—genom att inte titta på varje liten DNA‑skillnad, utan på ett fåtal regioner som fungerar som artspecifika streckkoder.

Problemet med liknande DNA
Får och getter delar mycket av sin genetiska mall, så korta DNA‑fragment från den ena arten passar ofta nästan lika bra mot den andras referensgenom. Författarna analyserade helgenomssekvenseringsdata från 40 individer med kända identiteter—20 får och 20 getter—var och en med hundratals miljoner läsningar. Med standardverktyg som matchar läsningar mot referensgenom fann de att båda arternas DNA alignade mycket väl mot både får‑ och getreferenserna. Aligneringsgrader, täckningsdjup och felmått var alla mycket lika och visade stor överlappning, vilket gjorde det nästintill omöjligt att med säkerhet avgöra vilken art ett prov kom ifrån baserat enbart på dessa rutinmässiga statistikmått.
Varför vanliga DNA‑klassificerare fallerar
Teamet testade också Kraken2, ett populärt program som försöker tilldela varje DNA‑läsning en plats i livets träd. Även med en omfattande databas klassificerades läsningar från både får och getter mestadels till samma breda djurgrupper, med endast små numeriska skillnader mellan dem. Visualiseringar av dessa tilldelningar visade att de flesta läsningar från båda arterna hamnade i samma genera, vilket speglar hur mycket av deras DNA de delar med varandra och med andra däggdjur. I praktiken innebär dessa suddiga gränser att traditionella taxonomiska verktyg kan vilseleda forskare som antar att en märkt ”får”‑dataset verkligen kommer från får, eller att ett felmärkt prov är lätt att upptäcka.
Att vända från täckning till artspecifika streckkoder
I stället för att fråga hur väl DNA‑läsningar matchar en referens vände författarna frågan: var matchar de inte? De alignade träningssetet på 30 djur (15 får, 15 getter) mot båda referensgenomen och skannade efter regioner med ett tydligt på–av‑mönster. En region räknades till exempel som ”get‑specifik” om getprover konsekvent visade normal täckning där när de alignades mot getgenomet, medan fårprover visade nästan ingen täckning på samma position. Med strikta tröskelvärden gav denna sökning mer än 150 000 kandidatregioner i getter och över 1,7 miljoner i får. Efter manuell granskning med fokus på längre, tydligt separerade sträckor destillerade teamet detta till bara tio högförtroende‑regioner per art—korta DNA‑zoner där en art pålitligt ”lyser upp” medan den andra förblir mörk.
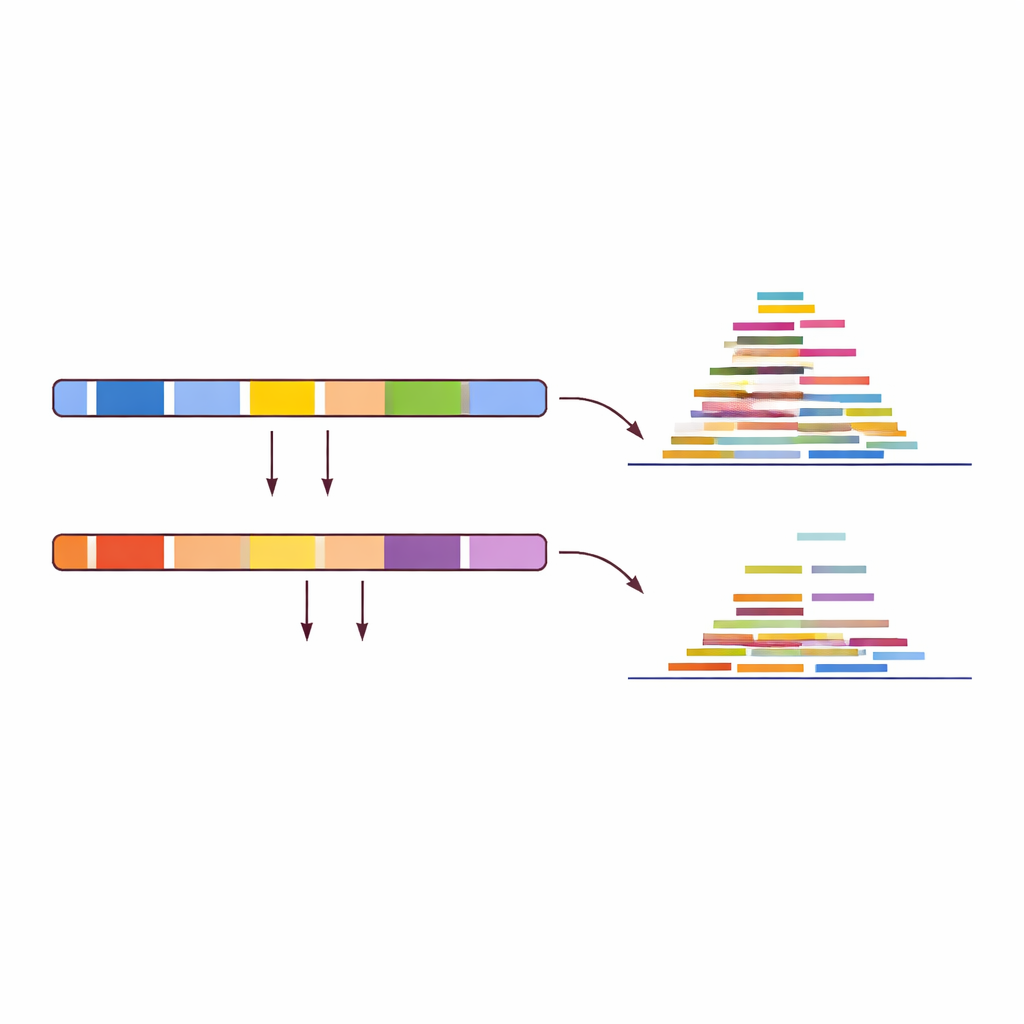
Ett enkelt test för okända prover
Med dessa 20 regioner i handen utformade författarna en rak testprocedur för valfri omärkt DNA‑dataset. Först alignas läsningarna mot både får‑ och getreferensgenomen. Sedan mäter man hur mycket täckning—ansamlingen av läsningar—som faller inom de tio får‑specifika regionerna i fårgenomet respektive de tio get‑specifika regionerna i getgenomet. Om fårregionerna visar stark täckning medan getregionerna nästan är tomma är provet ett får; om mönstret är omvänt är det en get. Tillämpat på 14 oberoende valideringsprover, inklusive offentligt tillgängliga data från olika sekvenseringsmaskiner och till och med kemiskt modifierat DNA, identifierade detta mönsterbaserade test varje enskilt prov korrekt och nådde 100 % noggrannhet i den studerade uppsättningen.
Nya verktyg och framtida användningsområden
Utöver att lösa ett praktiskt problem för får‑ och getforskning erbjuder detta arbete en generell mall som kan anpassas till andra par—eller grupper—av nära besläktade arter. De kurerade regionerna kan fungera som byggstenar för framtida verktyg, från snabba labbtester som amplifierar bara dessa artspecifika sträckor, till automatiserad programvara som skannar gamla sekvenseringsdatamängder för felmärkning. Även om metoden kräver att man alignar data mot flera referensgenom—vilket kostar beräkningstid och lagring—undviker den många fällor hos traditionella tillvägagångssätt och är robust mot skillnader i raser och sekvenseringsplattformar. I vardagliga termer har författarna visat hur ett mycket litet antal noggrant utvalda DNA‑landmärken kan ge ett klart, pålitligt svar på en fråga som stora, komplexa algoritmer ofta får fel på: vilket djur är detta?
Citering: dain Marzouka, N.a., Al-Aamri, A., Alshamsi, F. et al. A genomic approach for accurate identification of closely related species with next-generation sequencing samples. Sci Rep 16, 11329 (2026). https://doi.org/10.1038/s41598-026-41497-0
Nyckelord: artsidentifiering, helgenomssekvensering, får och getter, jämförande genomik, djurgenietik