Clear Sky Science · ar
نهج جينومي للتعرّف بدقّة على الأنواع المتقاربة باستخدام بيانات التسلسل من الجيل التالي
لماذا هذا مهم للمزارع وما بعدها
يمكن للتسلسل الحديث للحمض النووي قراءة الشفرة الجينية للحيوانات بتفصيل مدهش، لكن حتى الحواسيب القوية قد تتعثر عند سؤال بسيط نوعًا ما: هل هذه التسلسلات من خروف أم من معزة؟ بالنسبة للمزارعين والمربين والحماة والعلماء، فإن خلط الأنواع في مجموعات بيانات كبيرة للحمض النووي يمكن أن يعطل دراسات الصحة والإنتاجية والتطور. تقدم هذه الورقة طريقة بسيطة لكنها ذكية للتمييز بين الأنواع المتقاربة — مُوضّحة على الأغنام والماعز — من خلال عدم النظر إلى كل فرق صغير في حمضهم النووي، بل إلى عدد قليل من المقاطع التي تعمل كرموز شريطية خاصة بكل نوع.

المشكلة مع الحمض النووي المتشابه
تشترك الأغنام والماعز في جزء كبير من مخططهما الجيني، لذا غالبًا ما تتطابق لُقيم قصيرة من الحمض النووي من أحدهما تقريبًا بنفس القدر على جينوم النوع الآخر المرجعي. حلل المؤلفون بيانات التسلسل الجينومي الكامل من 40 حيوانًا معروفة هويتهم — 20 خروفًا و20 معزة — كل منها يتضمن مئات الملايين من قراءات الحمض النووي. باستخدام أدوات معيارية لمطابقة القراءات مع الجينومات المرجعية، وجدوا أن حمض كلا النوعين يتوافق بشكل جيد جدًا مع كل من المراجع الخاصة بالأغنام والماعز. كانت معدلات المحاذاة وعمق التغطية ومقاييس الأخطاء متشابهة جدًا وأظهرت تداخلاً كبيرًا، مما جعل من المستحيل تقريبًا أن تُحدَّد بثقة أي نوع جاء منه العينة بناءً على هذه الإحصاءات الروتينية وحدها.
لماذا تصنفات الحمض النووي الاعتيادية لا تكفي
اختبر الفريق أيضًا Kraken2، وهو برنامج شائع يحاول تعيين كل قراءة حمض نووي إلى موقع في شجرة الحياة. حتى مع قاعدة بيانات شاملة، تم تصنيف قراءات كل من الأغنام والماعز غالبًا ضمن نفس المجموعات الحيوانية العريضة، مع فروق رقمية طفيفة فقط بينهم. أظهرت تصورات هذه التعيينات أن معظم القراءات من كلا النوعين تتجمع في نفس الأجناس، مما يعكس مقدار الحمض النووي المشترك بينهما ومع الثدييات الأخرى. عمليًا، تعني هذه الحدود الضبابية أن الأدوات التصنيفية التقليدية قد تضِلّ الباحثين الذين يفترضون أن مجموعة بيانات معنونة «خروف» هي بالفعل من الخراف، أو أن العينة المُصنفة خطأ ستكون سهلة الاكتشاف.
الاستفادة من غياب التغطية لصياغة رمز شريطي نوعي
بدلًا من سؤال إلى أي مدى تتطابق القراءات مع مرجع، قلب المؤلفون السؤال: أين لا تتطابق؟ قاموا بمحاذاة مجموعة التدريب المكوّنة من 30 حيوانًا (15 خروفًا، 15 معزة) مع كلا الجينومين المرجعيين وفحصوا المناطق التي تظهر نمط تشغيل/إيقاف واضحًا. اعتبرت منطقة «خاصة بالماعز»، على سبيل المثال، إذا أظهرت عينات الماعز تغطية عادية هناك عند المحاذاة إلى جينوم الماعز، بينما أظهرت عينات الأغنام تقريبًا لا تغطية في نفس الموضع. باستخدام حدود صارمة، أنتج هذا البحث أكثر من 150 ألف منطقة مرشحة في الماعز وأكثر من 1.7 مليون في الأغنام. بعد مراجعة يدوية تركزت على المقاطع الأطول والمفصولة بوضوح، ركز الفريق هذا ليصل إلى عشرة مناطق عالية الثقة لكل نوع — مناطق قصيرة من الحمض النووي حيث يضيء نوع واحد بشكل موثوق بينما يظل الآخر مظلمًا.
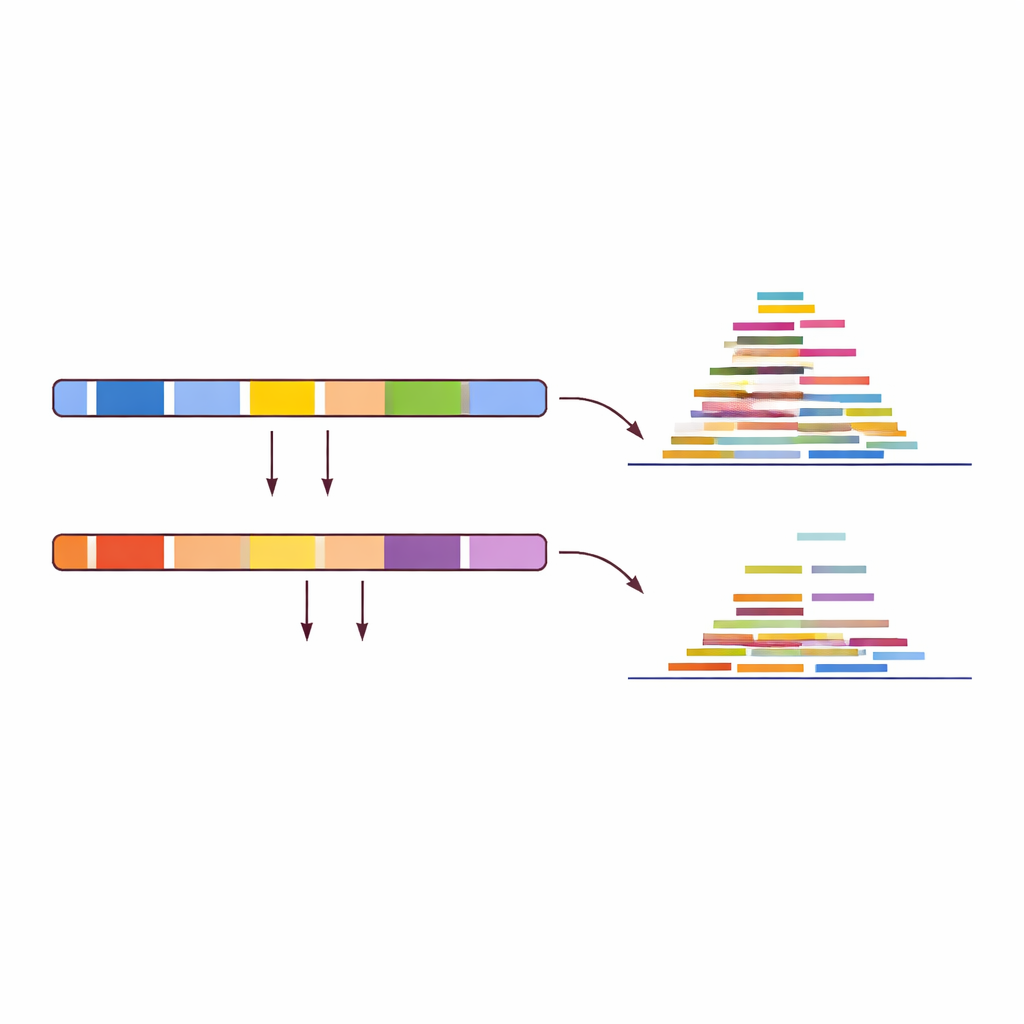
اختبار بسيط للعينات المجهولة
بوجود هذه المناطق العشرين، صمم المؤلفون روتين اختبار مباشر لأي مجموعة بيانات حمض نووي غير معنونة. أولًا، حرّك القراءات إلى كل من جينومي الأغنام والماعز المرجعيين. ثم، قِس مقدار التغطية — تراكُم القراءات — داخل العشر مناطق الخاصة بالأغنام على جينوم الأغنام والعشر مناطق الخاصة بالماعز على جينوم الماعز. إذا أظهرت مناطق الأغنام تغطية قوية بينما كانت مناطق الماعز شبه فارغة، فالعينة خروف؛ وإذا كان النمط معكوسًا، فهي معزة. عند تطبيق هذا الاختبار المعتمد على النمط على 14 عينة تحقق مستقلة، بما في ذلك بيانات متاحة علنًا من آلات تسلسل مختلفة وحتى حمض نووي معدل كيميائيًا، حدّد الاختبار كل عينة بشكل صحيح، محققًا دقة 100% في المجموعة المدروسة.
أدوات جديدة واستخدامات مستقبلية
بعيدًا عن حل مشكلة عملية لأبحاث الأغنام والماعز، يقدم هذا العمل مخططًا عامًا يمكن تكييفه لأزواج — أو مجموعات — أخرى من الأنواع المتقاربة. تعمل المناطق المنقّحة كلبنات بناء لأدوات مستقبلية، من اختبارات مخبرية سريعة تضخم فقط تلك المقاطع الخاصة بكل نوع، إلى برامج آلية تفحص مجموعات بيانات التسلسل القديمة لاكتشاف الوسم الخاطئ. على الرغم من أن الطريقة تتطلب محاذاة البيانات إلى جينومات مرجعية متعددة، مما يستهلك وقت حوسبة ومساحة تخزين، إلا أنها تتجنب العديد من مطبات النهج التقليدية وتتحمّل الاختلافات بين السلالات ومنصات التسلسل. ببساطة يومية، أظهر المؤلفون كيف أن عددًا صغيرًا من المعالم الجينية المختارة بعناية يمكن أن يعطي إجابة واضحة وموثوقة على سؤال غالبًا ما تجيب عنه الخوارزميات الكبيرة والمعقدة بشكل خاطئ: أي حيوان هذا؟
الاستشهاد: dain Marzouka, N.a., Al-Aamri, A., Alshamsi, F. et al. A genomic approach for accurate identification of closely related species with next-generation sequencing samples. Sci Rep 16, 11329 (2026). https://doi.org/10.1038/s41598-026-41497-0
الكلمات المفتاحية: تحديد الأنواع, التسلسل الجينومي الكامل, الأغنام والماعز, علم الجينوم المقارن, علم وراثة الحيوانات