Clear Sky Science · sv
AI-baserad fragmentoptimering av sorafenib-derivat riktade mot VEGFR2 för angiogenesrelaterade sjukdomar: en struktur‑baserad in silico‑studie
Varför omformning av ett cancerläkemedel spelar roll för många sjukdomar
Nya blodkärl håller våra organ vid liv, men när deras tillväxt går ur kontroll kan de försörja tumörer, skada ögat och störa moderkakan under graviditet. Denna studie undersöker hur artificiell intelligens och avancerade datorsimuleringar kan omdesigna ett befintligt cancerläkemedel, sorafenib, för att mer precist kontrollera en nyckelregulator för blodkärlstillväxt kallad VEGFR2, med långsiktig förhoppning om säkrare behandlingar för angiogenesrelaterade sjukdomar.
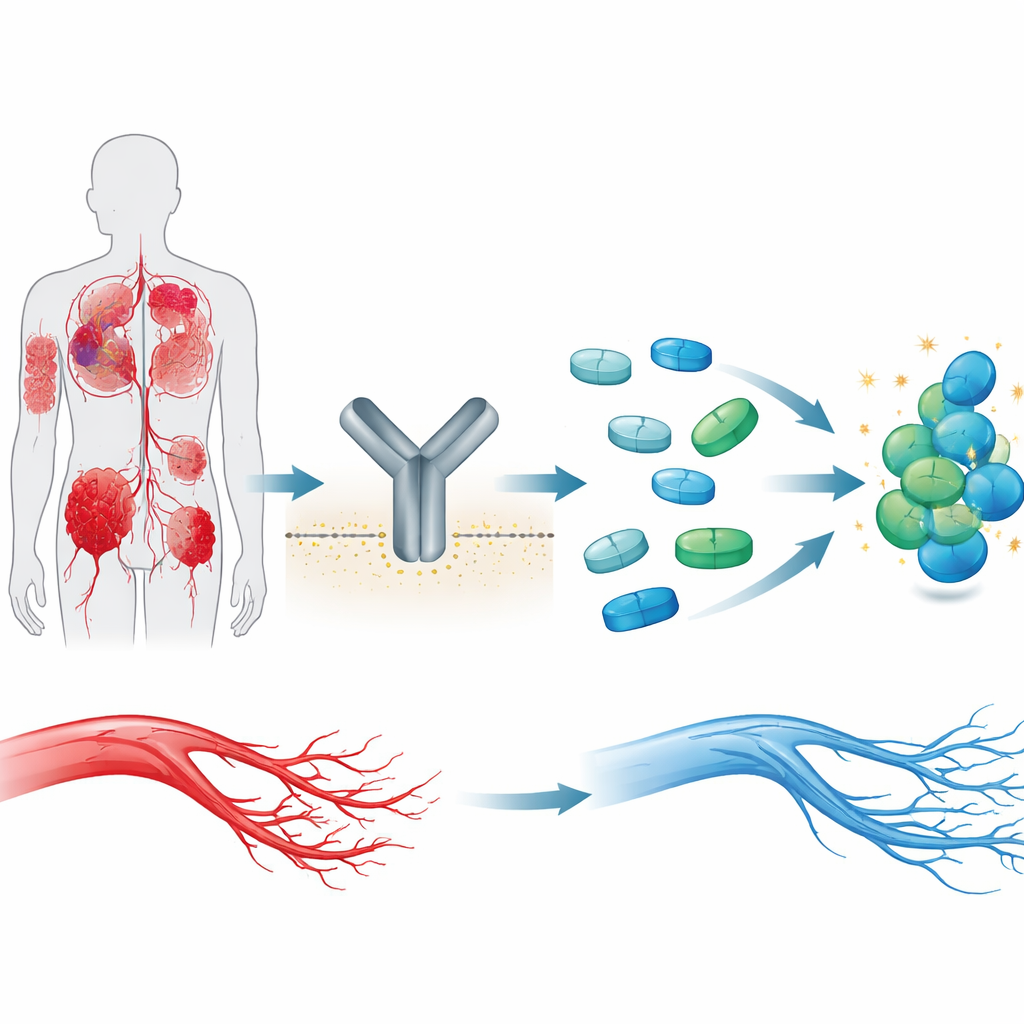
Rödljuset och grönt ljus för nya blodkärl
Blodkärlstillväxt, eller angiogenes, är i hög grad beroende av ett protein på ytan av kärlbeklädnadsceller kallat VEGFR2. När dess partnermolekyl VEGF binder aktiveras VEGFR2 och signalerar åt cellerna att dela sig, migrera och bilda nya kärl. Det är användbart vid sårläkning och utveckling, men skadligt när det driver tumörers blodförsörjning, vissa ögonsjukdomar, kronisk inflammation eller onormala förändringar i moderkakan. På grund av denna centrala roll har VEGFR2 blivit ett stort läkemedelsmål, och flera läkemedel blockerar redan denna receptor. Många av dessa läkemedel, inklusive sorafenib, påverkar dock flera besläktade proteiner, vilket kan leda till allvarliga biverkningar.
Att förvandla sorafenib till ett bättre verktyg
Sorafenib är ett kraftfullt men grovhugget verktyg. Det binder till VEGFR2:s inre ”motor” och liknande enzymer och släcker signaler som främjar blodkärlstillväxt. Tyvärr medför dess breda aktivitet problem som förhöjt blodtryck, hjärtpåfrestning, hudreaktioner och risk för leverskada. Istället för att börja från början använde författarna sorafenibs grundläggande kemiska skelett som mall och frågade: kan vi använda AI för att varsamt omforma denna molekyl så att den binder VEGFR2 tätare, beter sig bättre i kroppen och ser mindre toxisk ut—utan att förlora sin anti‑angiogena effekt?
Låta AI utforska säkrare kemiskt utrymme
Gruppen matade in sorafenibs struktur i ett AI‑drivet system som gör små, kemiskt realistiska förändringar—lägger till eller byter ut fragment på sätt som en läkemedelskemi skulle kunna återskapa i labbet. Detta gav tjugo nya kandidatmolekyler som alla behöll sorafenibs kärnegenskaper men skilde sig i sidogrupper och övergripande form. Forskarna förlitade sig sedan helt på datorbaserade metoder för att pröva dessa kandidater: virtuell dockning mot en högupplöst 3D‑modell av VEGFR2, långa molekyldynamik‑simulationer som efterliknar atomernas rörelse i vattenlika, kroppslika förhållanden, och energiberäkningar som uppskattar hur starkt varje molekyl binder.
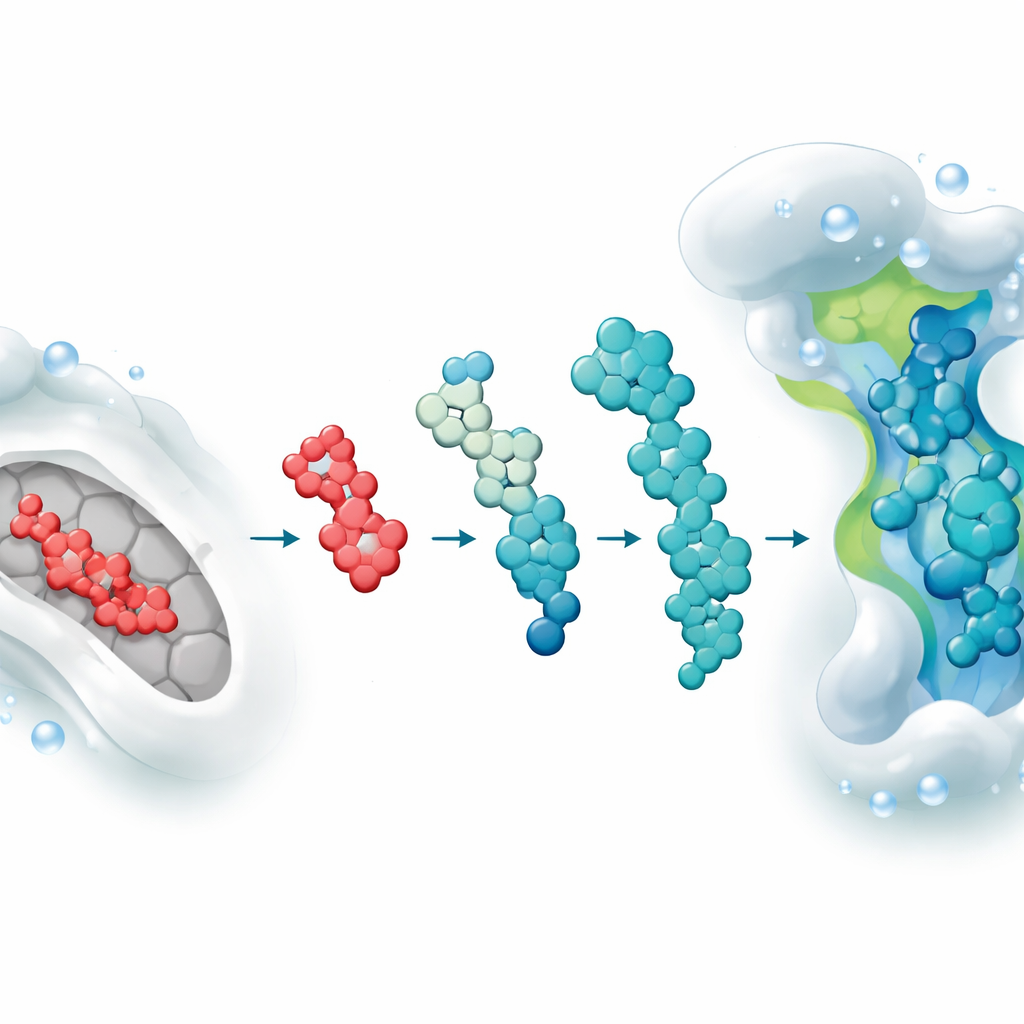
En framstående kandidat i simuleringarna
En design, kallad AI‑Fragmented Derivative 7 (Grow), steg konsekvent fram som bäst. Dockningsstudier antydde att den slog sig ner i VEGFR2:s huvudsakliga aktiva spår mer tätt än sorafenib och bildade ett tätare nät av kontakter med viktiga aminosyror som är kända för att vara betydelsefulla för verkliga hämmare. En halv mikrosekund av molekyldynamik‑simulationer visade att komplexet förblev förvånansvärt stabilt över tid: proteinet höll sig kompakt, bindningsfickan kollapsade eller deformeras inte, och den nya molekylen drev knappt utan bibehöll flera vätebindningar och hydrofoba kontakter genom hela simuleringen. Avancerade kvantmekaniska beräkningar indikerade att Derivative 7:s elektronfördelning gynnar laddningsinteraktioner med VEGFR2, vilket stöder dess förutspådda starka bindning.
Tecken på bättre beteende i kroppen
Utöver att binda väl måste ett läkemedel röra sig säkert i kroppen. Författarna använde webbaserade prediktionsverktyg för att uppskatta hur varje kandidat troligen skulle lösa sig, absorberas, brytas ned av leverenzym och potentiellt orsaka skada. Derivative 7 förväntades ha något bättre vattenlöslighet och en mer balanserad blandning av oljiga och polära regioner än sorafenib—egenskaper som ofta underlättar dosering och formulering. Viktigt var att maskininlärningsbaserade toxicitetsmodeller flaggade sorafenib som mer sannolik att orsaka läkemedelsinducerad leverskada, medan Derivative 7 föll på den säkrare sidan av denna risk. Samtidigt behöll den en i stort sett liknande metabol profil, vilket tyder på att dess nedbrytningsvägar kan bli förutsägbara.
Från datorlöften till verklig bevisning
I vardagstermer visar detta arbete hur AI och fysikbaserade simuleringar kan ”hyvla och polera” ett befintligt läkemedel till en potentiellt skarpare och säkrare version riktad mot samma molekylära brytare. Det omdesignade sorafenib‑derivatet verkar i beräkningarna binda VEGFR2 mer bestämt och bära färre varningssignaler för leverskada, samtidigt som de grundläggande egenskaperna som gör sorafenib effektivt bevaras. Alla dessa fynd är dock förutsägelser: den nya molekylen har ännu inte syntetiserats eller testats i celler, djur eller patienter. Studiens verkliga bidrag är att erbjuda en välmotiverad, datafylld kandidat—och en återanvändbar digital pipeline—för nästa generations läkemedel som bättre kan tygla okontrollerad blodkärlstillväxt vid cancer, ögonsjukdomar och andra angiogenesdrivna tillstånd.
Citering: Inan, D., Karageçili, S. & Ali, N. AI fragmentation-based optimization of Sorafenib derivatives targeting VEGFR2 for angiogenesis-related pathologies: a structure-based in-silico study. Sci Rep 16, 11848 (2026). https://doi.org/10.1038/s41598-026-41232-9
Nyckelord: angiogenes, VEGFR2‑hämmare, sorafenib‑derivat, AI‑läkemedelsdesign, molekylär dockning