Clear Sky Science · sv
Atomistisk modellering av molekylära interaktioner med kopparoxider för korrosionshämning
Varför koppar behöver osynliga livvakter
Koppar driver tyst mycket av det moderna livet, från kretskort och bilens elektronik till vattenrör och värmeväxlare. Men i luft som innehåller fukt och föroreningar korroderar koppar långsamt och tappar prestanda. Denna översiktsartikel förklarar hur forskare använder datorsimuleringar för att förstå och förbättra tunna molekylära filmer som skyddar koppar från denna tysta skada, med särskilt fokus på de realistiska, röriga ytor som bildas i verkligheten snarare än på idealiserade labbmodeller.
Vanligt metall och dess dolda svaghet
Koppar är populärt eftersom det leder värme och elektricitet väl och är lätt att forma. Men så fort det exponeras för luft börjar dess blanka rosa yta reagera med syre och vatten. Ett tunt rött skikt av kopparoxid bildas, vilket till en början bromsar ytterligare angrepp. Med tiden störs dock detta skyddsskikt av fukt, salt och andra föroreningar. Oxiden förtjockas, defekter uppstår och nya grönaktiga korrosionsprodukter kan växa ovanpå. Dessa förändringar kan minska ledningsförmågan hos tunna kopparfolier som används i elektronik och lämna industrikomponenter sårbara för fel.
Hur skyddande molekyler bygger en sköld
För att begränsa korrosion applicerar ingenjörer speciella oorganiska salter eller organiska molekyler som fäster vid koppar och bildar en barriärfilm. Många framgångsrika organiska hämmare, såsom azoler och närbesläktade föreningar, innehåller atomer som kväve eller svavel som kan dela elektroner med kopparatomer. De sitter på ytan antingen svagt eller starkt och kan montera sig till ett tätt, ordnat skikt som blockerar vatten och aggressiva joner från att nå metallen. Experiment visar till exempel att 2-mercaptobenzimidazol och liknande molekyler kan bilda självmonterande monolager på koppar som fungerar i både sura och salta lösningar.
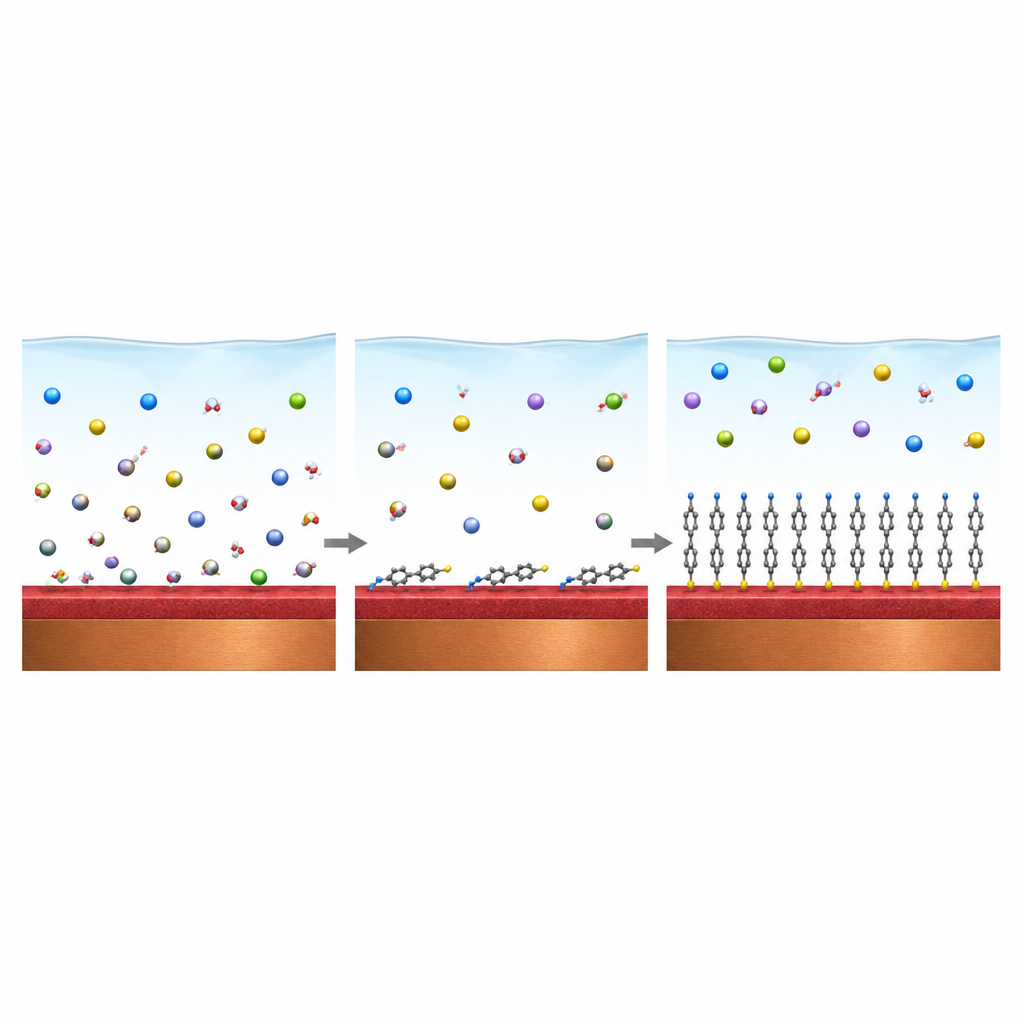
Varför realistiska ytbilder spelar roll
De flesta datorstudier har behandlat koppar som en perfekt ren, plan metallyta. I verkligheten är koppar normalt täckt av ett eller flera oxidskikt som är grova, trappstegsformade och ibland delvis brutna, särskilt när kloridjoner är närvarande. Denna översikt samlar arbeten som rör sig bort från den enkla bilden. Forskare modellerar nu kopparoxidskivor av olika tjocklek, ibland stödda av en kopparbas, ibland med vakanser, steg och lokalt nakna fläckar. De undersöker också hur vattenlager och upplösta saltjoner ligger ovanpå dessa oxider och konkurrerar med hämmarmolekyler om samma bindningsställen.
Att skåda korrosion med digitala mikroskop
Flera nivåer av simulering används. Klassisk molekylär dynamik behandlar atomer som interagerande kulor och kan köras under långa tider för att visa hur vatten, joner och hämmare rör sig nära ytan, men den kan inte hantera förändringar i elektrondistribution som ligger till grund för kemisk bindning. Densitetsfunktionalteori, en kvantmetod, ger detaljerad information om föredragna bindningsställen, bindningsstyrkor och laddningsöverföring mellan molekyler och kopparoxider, men är begränsad till mindre system och korta tider. Hybridmetoder och nyare maskininlärningsmodeller syftar till att kombinera kvantmetoders noggrannhet med storskalig dynamiks räckvidd, och kan till och med börja inkludera effekten av applicerad spänning, vilket är avgörande i verklig elektrolytisk korrosion.
Öppna frågor och framtida verktyg
Trots framsteg kvarstår viktiga luckor. Många nuvarande modeller använder fortfarande oxidskikt som är för tunna, ignorerar den lätta tiltning som kan finnas mellan oxid- och metallkristaller i experiment, eller inkluderar inte fullt ut bulkvatten och upplösta joner. Viktigast är att mycket få simuleringar tar hänsyn till den elektriska potential som driver korrosionsreaktioner under driftförhållanden. Författarna argumenterar för att mer realistiska kopparoxidy tor, explicita vätskelager med salt och noggrann behandling av elektrodpotential är nödvändiga för att förutsäga hur hämmande filmer bildas, omorganiseras och ibland misslyckas. De framhåller lovande vägar såsom hybridkvant–klassiska scheman och maskininlärningspotentialer utformade för koppar, dess oxider, vatten och hämmarmolekyler.
Vad detta betyder för att skydda koppar
För icke-specialister är huvudbudskapet att datormodeller blir tillräckligt kraftfulla för att visa, atom för atom, hur skyddande molekyler tränger bort vatten och salt för att fästa vid kopparoxider och bromsa korrosion. Genom att göra dessa modeller närmare verkliga förhållanden hoppas forskare kunna förklara varför vissa hämmare fungerar bättre än andra och vägleda utformningen av säkrare, mer effektiva föreningar. På lång sikt kan denna djupare förståelse hjälpa till att hålla kopparn i våra apparater, fordon och infrastrukturer fungerande pålitligt längre, även i tuffa miljöer.
Citering: Iqbal, M., Martins, E.d.F., Todorova, N. et al. Atomistic modeling of molecular interactions with copper oxides for corrosion inhibition. npj Mater Degrad 10, 62 (2026). https://doi.org/10.1038/s41529-026-00779-8
Nyckelord: kopparkorrosion, korrosionshämmare, kopparoxider, molekylär modellering, självmonterande monolager