Clear Sky Science · sv
Upptäckt av högentropiska fasta elektrolyter: ett tvåstegs maskininlärningsramverk som kopplar atomära konfigurationer till jontransportegenskaper
Varför denna forskning är viktig
Moderna elbilar och bärbara enheter förlitar sig i allt högre grad på fasta batterier som är säkrare och mer kompakta än dagens vätskebaserade konstruktioner. En nyckelkomponent i detta pussel är den fasta elektrolyten, materialet som låter litiumjoner röra sig mellan batteriets elektroder. Denna artikel beskriver ett nytt datorstyrt sätt att söka bland tusentals komplexa elektrolytrecept för att hitta dem som låter joner färdas snabbt, vilket kan hjälpa ingenjörer att utforma bättre fasta batterier mycket mer effektivt.

Utmaningen med trånga kemiska recept
Forskare har upptäckt att ”högmentropiska” elektrolyter, där många olika grundämnen delar atomiska platser slumpmässigt, ibland kan leda litiumjoner mycket väl. Men denna kemiska rikedom har sitt pris. Antalet möjliga kombinationer exploderar, och traditionella försök-och-fel-experiment eller långsamma kvantberäkningar kan inte realistiskt testa dem alla. I dessa täta strukturer ändrar små förskjutningar i atompositioner hur lätt joner kan slingra sig genom materialet, vilket gör det svårt att i förväg gissa vilka recept som fungerar bäst.
Användning av smarta modeller som genväg
Författarna tacklar denna utmaning med ett tvåstegs ramverk för maskininlärning byggt kring en välkänd fast elektrolyt kallad LZSP. I det första steget finjusterar de en befintlig neuronnäts-potential, CHGNet, så att den kan efterlikna kostsamma kvantberäkningar för denna materialfamilj. Denna anpassade modell relaxerar snabbt atomära strukturer och kör virtuella upphettningstester som följer hur litiumjoner vandrar över tid. Den når en noggrannhet nära betrodda kvantmetoder samtidigt som beräkningstiden minskas från dagar till timmar eller mindre.
Att koppla struktur till jonrörelse
Det andra steget omvandlar problemet till något ännu snabbare. Istället för att simulera varje kandidat i detalj tränar forskarna en separat modell som länkar enkla strukturella egenskaper till hur långt litiumjoner tenderar att förflytta sig. De matar in kvantiteter som hur många litiumatomer som finns, hur förvrängda vissa atomkaféer är och hur utdragen kristallcellen är. Modellen lär sig vilka mönster som hör ihop med trög rörelse och vilka som hör ihop med långa jonhopp. Med denna genväg kan teamet snabbt uppskatta jonmobilitet för tusentals hypotetiska material utan att köra fullständiga simuleringar varje gång.
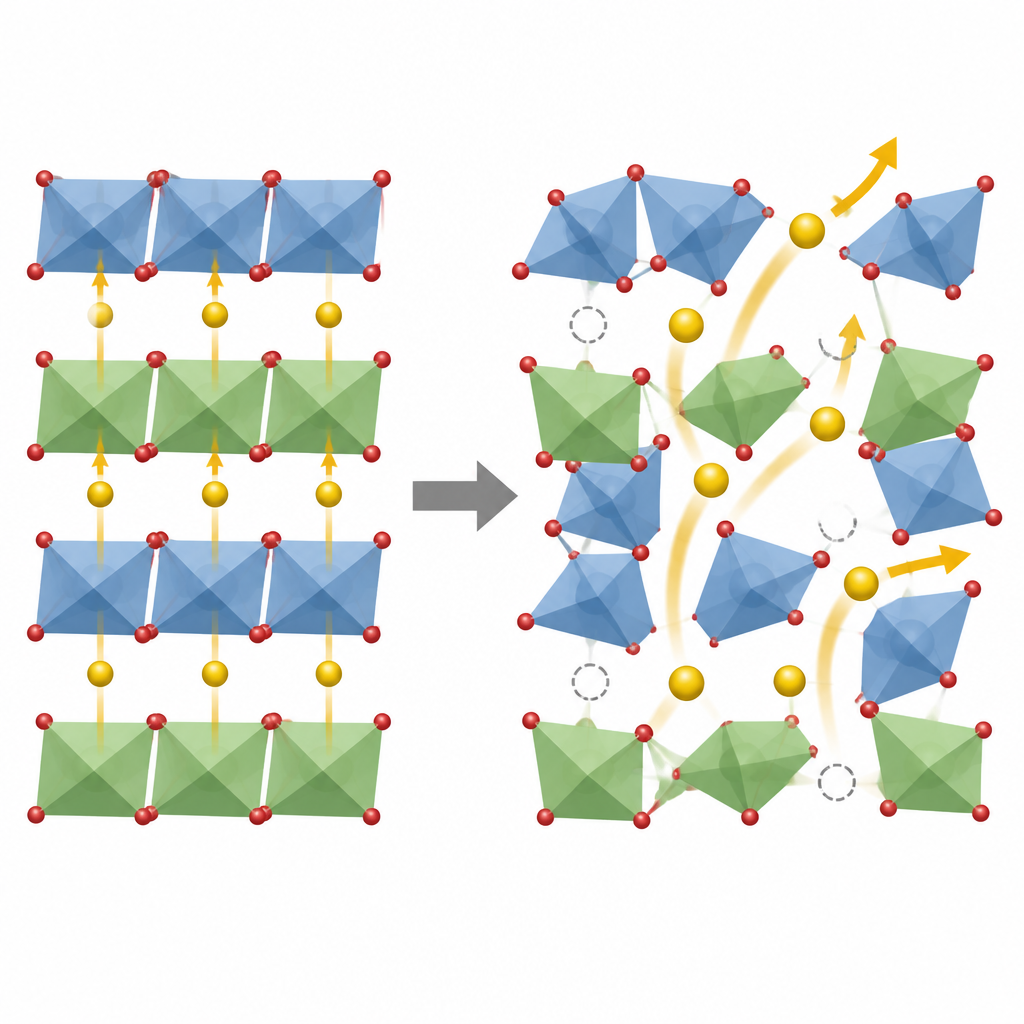
Att hitta framstående fasta elektrolyter
Beväpnade med denna tvåstegsstrategi skannar forskarna ett stort utrymme av femkomponentsversioner av LZSP, som täcker 4575 olika sammansättningar. Deras funktionsbaserade modell fungerar som ett filter och rankar kandidater efter förväntad jonmobilitet. De tillämpar sedan den mer detaljerade simuleringen endast på de högst rankade få. Denna pipeline avslöjar en särskild blandning av zirkonium, hafnium, tenn, titan och niob som förutsägs leda litiumjoner ungefär tusen gånger bättre vid rumstemperatur än den ursprungliga LZSP. Beräkningarna visar också varför: rätt kombination av grundämnen skapar litiumvakanser och milda förvrängningar i det atomära ramverket som öppnar sammanhängande, lågresistiva vägar för jonflöde samtidigt som gitterstrukturen förblir stabil.
Vad fynden betyder för framtida batterier
För icke-specialister är huvudbudskapet att författarna har byggt en smart sil för batterimaterial. Istället för att kontrollera varje möjligt recept med långsamma, detaljerade beräkningar eller laboratorieexperiment använder de snabba, tränade modeller för att sortera bort dåliga presterande alternativ och lyfta fram en liten grupp värd noggrann uppmärksamhet. Detta tillvägagångssätt pekar inte bara ut en särskilt lovande kandidat till fast elektrolyt, utan klargör också vilka strukturella drag som tenderar att främja snabb jonrörelse. Eftersom metoden är generell kan den anpassas till andra fasta elektrolyter och till och med andra egenskaper, vilket erbjuder en praktisk färdplan för att utforska enorma kemiska rum på ett målinriktat och tidsbesparande sätt.
Citering: Fu, X., Xu, J., Yang, Q. et al. High-entropy solid electrolytes discovery: a dual-stage machine learning framework bridging atomic configurations and ionic transport properties. npj Comput Mater 12, 178 (2026). https://doi.org/10.1038/s41524-026-02041-w
Nyckelord: fasta elektrolyter, högmentropiska material, litiumjontransport, maskininlärning för material, fasta batterier