Clear Sky Science · sv
Kemiska bindningskoncept framträder naturligt ur maximalt intrasslade atomorbitaler
Varför denna nya syn på kemiska bindningar spelar roll
Läroböcker i kemi lär oss att föreställa oss bindningar som enkla linjer mellan atomer, men riktiga molekyler beter sig enligt kvantfysikens märkliga lagar. Denna artikel visar hur idéer från kvantinformation, särskilt hur starkt olika delar av ett system är kopplade, kan ge en tydlig och kvantitativ bild av kemiska bindningar. Arbetet knyter de bekanta klassrumsskisserna av molekyler till elektronernas djupare kvantstruktur och erbjuder ett enhetligt sätt att tänka på vanliga bindningar, flercentriska bindningar och aromatiska ringar.
Ett nytt sätt att se hur atomer håller ihop
Kemiska bindningar beskrivs vanligtvis med två klassiska bilder. Valensbindningsteori fokuserar på elektronpar som delas mellan atomer, medan molekylorbitalteori sprider elektroner över hela molekylen. Moderna datormetoder kan förutsäga energier mycket noggrant, men de döljer ofta den enkla bindningsberättelsen bakom lager av matematisk detalj. Författarna föreslår en annan väg. De utgår från lokaliserade atomorbitaler, använder verktyg från kvantinformation för att mäta hur starkt dessa orbitaler är kopplade, och återfår därigenom de bekanta bindningsmönstren som kemister ritar för hand.
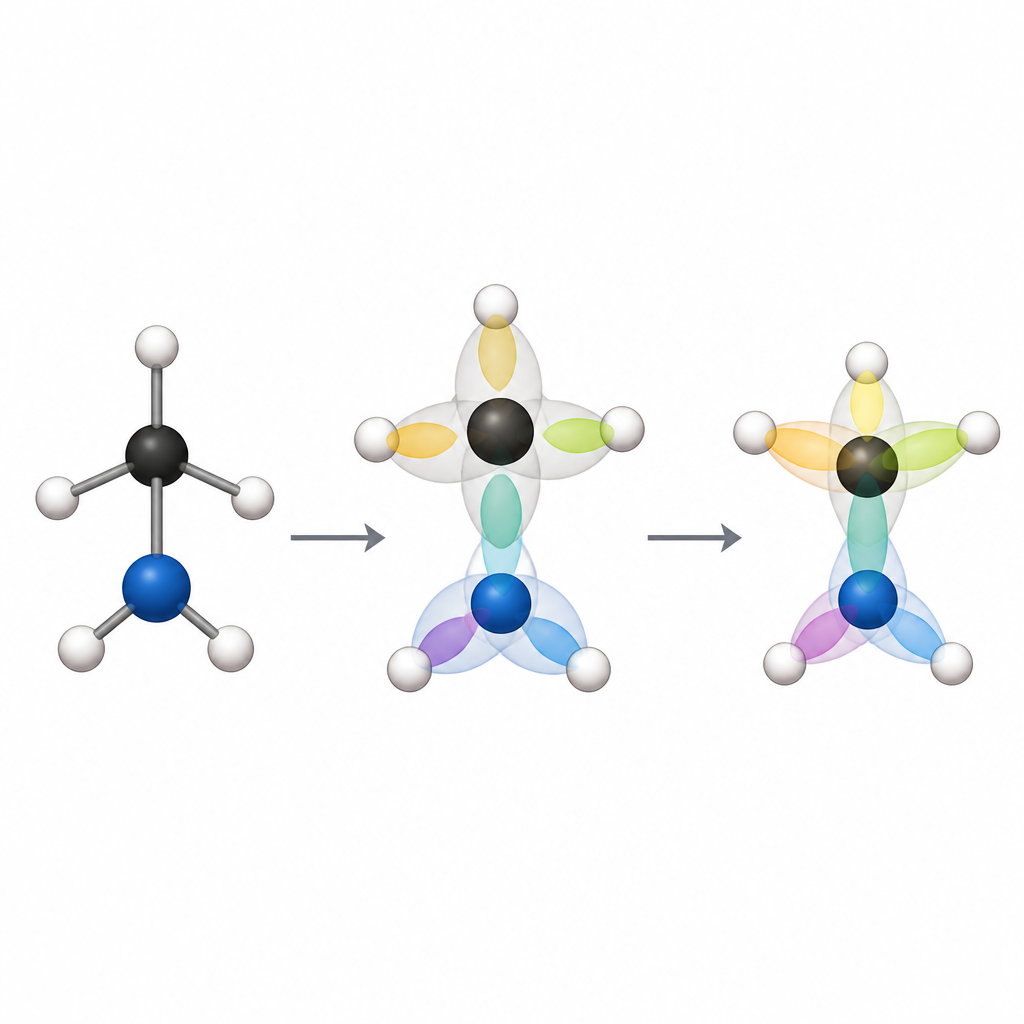
Maximalt intrasslade atomorbitaler i enkla termer
Den centrala idén är en särskild uppsättning lokaliserade orbitaler kallade maximalt intrasslade atomorbitaler. Här betyder "intrasslad" att vad som händer med elektroner i en orbital är tätt kopplat till vad som händer i en annan orbital, på ett sätt som bara kvantmekaniken tillåter. Författarna väljer och roterar startorbitalerna så att den totala kopplingen mellan orbitaler på olika atomer blir så stark som möjligt. När de sedan undersöker hur par eller grupper av dessa orbitaler korrelerar, finner de att varje starkt par motsvarar en konventionell bindning, och grupper om mer än två orbitaler avslöjar mer komplexa bindningsmönster.
Återfå bekanta bindningar och följa bindningsstyrka
Genom att testa sin metod på enkla molekyler visar forskarna att dessa speciella orbitaler automatiskt återger välkända kemiska egenskaper. I eten, till exempel, omorganiserar kolorbitalerna sig till det bekanta sp2-mönstret utan några inbyggda kemiska regler. Starkt kopplade orbitalpar kartläggs ett-till-ett på enkla, dubbla och tredubbla bindningar, och mängden kvantkoppling i ett par följer nära den vanliga idén om bindningsordning. Kovalenta bindningar visar hög intrassling, medan mer joniska eller svagt bundna system som litiumfluorid och heliumdimern uppvisar mycket lägre eller till och med försvinnande värden. Metoden fångar också subtila fall som "harpun"-mekanismen i litiumhydrid, där graden av delning mellan atomer först ökar och sedan minskar när bindningen töjs ut — något som standardpopulationanalyser har svårt att beskriva.
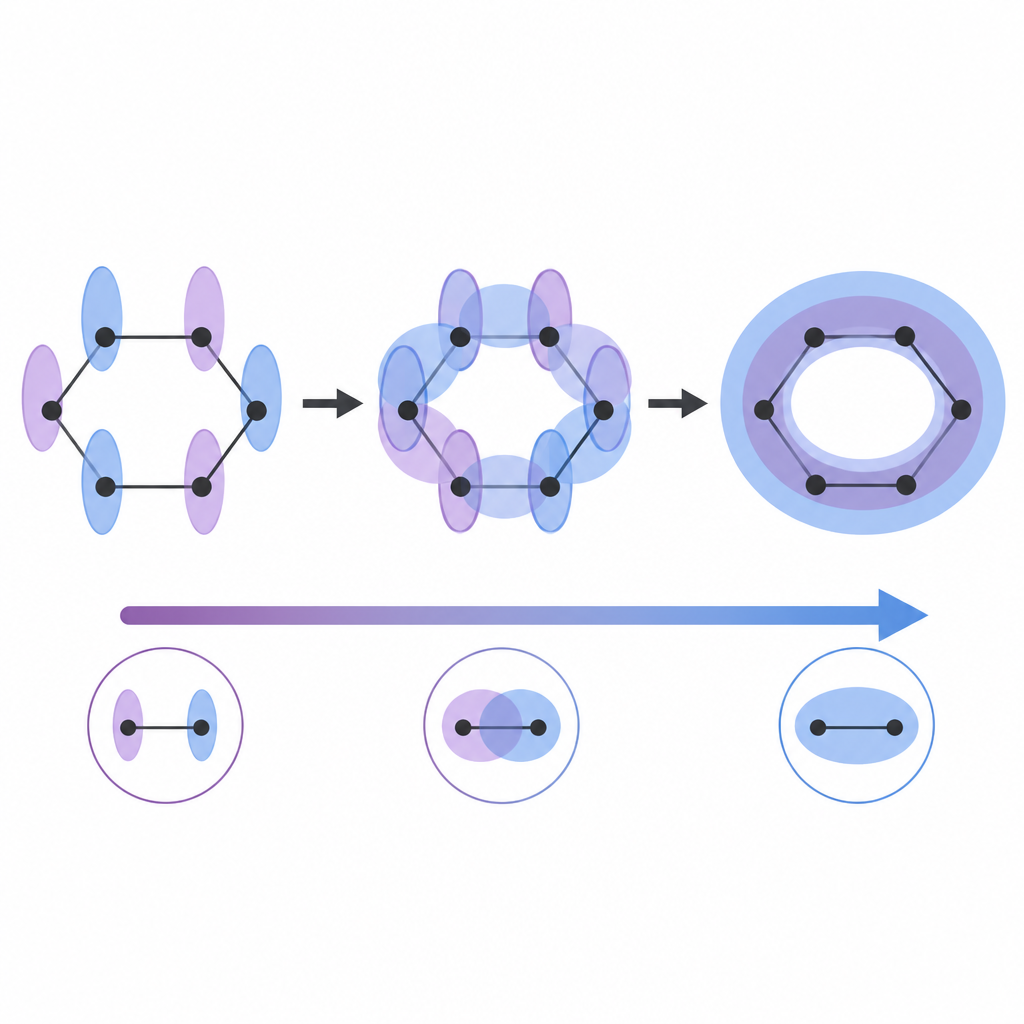
Se flercentriska bindningar och aromatiska ringar som delade mönster
Många molekyler kan inte beskrivas av enkla tvåatomiga bindningar. Författarna utvidgar sitt tillvägagångssätt genom att se hur intrassling delas mellan tre eller fler orbitaler samtidigt, en egenskap känd som genuin multipartit intrassling. I trecentersbindningar och i kluster av metall- och huvudgrupps-atomer signalerar hög multipartit intrassling att elektroner är utspridda över flera atomer på ett koordinerat sätt. Aromatiska molekyler ger ett ännu rikare test. I bensen bildar sex utanför-plan-orbitaler en starkt sammanlänkad ring med ett mycket högt värde av multipartit intrassling, vilket återspeglar den klassiska bilden av elektroner som cirkulerar runt ringen. När några kol byts ut mot kväve eller när ringen deformeras, sjunker detta värde, i linje med den vedertagna idén att aromatisk karaktär minskar under dessa förändringar.
Från läroboksbilder till en enhetlig kvantberättelse
Tillsammans visar resultaten att ett enda ramverk baserat på kvantinformation kan beskriva vanliga bindningar, flercentriska bindningar, aromatiskhet och till och med knepiga övergångstillstånd i reaktioner. Istället för att förlita sig på flera separata bindningsmodeller kan kemister i princip avläsa bindningsstyrka och bindningsmönster direkt ur hur starkt lokaliserade orbitaler är kopplade i elektronernas kvanttillstånd. För en lekman är huvudbudskapet att linjerna och ringarna som ritas i kemiska strukturer inte bara är praktiska symboler; de speglar djupa mönster av kvantkoppling som denna nya metod nu kan kvantifiera på ett precist och systematiskt sätt.
Citering: Ding, L., Matito, E. & Schilling, C. Chemical bonding concepts emerge naturally from maximally entangled atomic orbitals. Nat Commun 17, 4732 (2026). https://doi.org/10.1038/s41467-026-73527-w
Nyckelord: kemisk bindning, kvantintrassling, aromatiska egenskaper, flercentersbindningar, molekylorbitaler